 Lili Lou1,2
Lili Lou1,2 Hui Guo
Hui Guo Meiying Shao
Meiying Shao- 1Department of Pediatric Pulmonology and Immunology, West China Second University Hospital, Sichuan University, Chengdu, China
- 2Key Laboratory of Birth Defects and Related Diseases of Women and Children (Sichuan University), Ministry of Education, Chengdu, China
- 3Department of Pediatrics, West China Second University Hospital, Sichuan University, Chengdu, Sichuan, China
- 4West China School of Public Health and West China Fourth Hospital, Sichuan University, Chengdu, Sichuan, China
Background: Systemic lupus erythematosus is an autoimmune disease with diverse clinical manifestations. The symptoms of SLE in children are more atypical than adults. Childhood SLE complicated with Fanconi syndrome is extremely rare and even more difficult to diagnose.
Case presentation: This article reports a preschool boy with SLE who presented with renal tubular acidosis, accompanied by weakness in both lower limbs, delayed growth, and malnutrition. It was later found that the patient had the complication of Fanconi syndrome with renal tubular acidosis. Ultimately, renal biopsy confirmed lupus nephritis. The patient was treated with corticosteroid combined with mycophenolate mofetil, hydroxychloroquine, and belimumab. The symptoms of the child were relieved.
Conclusion: Here we report an extremely rare case of childhood SLE complicated with Fanconi syndrome. There has been no similar clinical report. It is necessary to be alert to the possibility of atypical SLE in children to avoid missed diagnosis and misdiagnosis.
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease that involves multiple organs and systems throughout the body. Multiple auto-antibodies, represented by antinuclear antibodies (ANA), exist in SLE patients, which can cause irreversible damage to affected organs. Lupus nephritis (LN) is the most common and important complication of SLE, and also one of the main causes of death in patients with SLE. In recent years, a large number of studies have shown that the pathogenesis of childhood-onset SLE (cSLE) is the result of the interaction among genetic, immunological, racial, and environmental factors. The prevalence rate of cSLE is approximately 3.3–24 per 100,000, while only 10%–20% of the patients are diagnosed during childhood (1, 2). The clinical manifestations of cSLE are diverse, with 60%–85% of children having atypical onset symptoms, and some children only showing slight abnormalities in urine routine tests in the early stage, which poses a difficulty and interference to clinical diagnosis (3, 4). Childhood lupus nephritis often involves the glomerulus, renal tubules, and interstitium, while the involvement of the latter two is rare (5). Compared with adult SLE, cSLE progresses more rapidly, has a wider range of involvements, is more severe, and has a poorer prognosis and a higher incidence of renal damage (1). About 5%–22% of children with LN will develop end-stage renal disease (ESRD) within 5–10 years (6–9). Therefore, early diagnosis and treatment are crucial for reducing organ damage, improving prognosis, and reducing mortality. This article reports a boy with preschool onset of SLE, with presenting symptoms of renal tubular acidosis such as fatigue, polyuria, polydipsia, and delayed growth and development. During the course of the disease, the complication of Fanconi syndrome was found. Except for positive auto-antibodies, there were no typical lupus symptoms such as fever, rash, alopecia, arthritis, and oral ulcers. Finally, renal biopsy confirmed lupus nephritis. Therefore, this case of cSLE had extremely atypical clinical manifestations.
Case presentation
A 4-year- and 8-month-old boy was brought to our hospital with fatigue, polyuria, and excessive drinking for one month. There was no recent history of fever, hair loss, facial erythema, oral ulcers, vomiting or diarrhea. There was no consanguineal marriage history, no family history of kidney disease and other genetic diseases. He was born spontaneously at 39 weeks, weighed 2.8 kg at birth, was 50 cm long, and had no history of asphyxia (Apgar score unknown). The family thought that he was shorter than other children of the same age and gender, but he was not lagging behind in motor, language and intellectual development. At the age of 1, he was 72 cm tall (−2SD∼−1SD) and weighed 8.5 kg (−2SD∼−1SD); he was 103 cm tall and weighed 12 kg at the time of consultation, and his physical examination height and weight were less than 2 standard deviations of the standard value, his intellectual development was normal, and he could talk and express needs normally. Blood pressure was normal, and there was no edema throughout the body except a ∼3 cm × 3 cm area of cafe au lait spoton the skin of the left lower limb. No other abnormalities were found on physical examinations. Urinary examination showed alkaline urine (urine pH >5.5), glycosuria and highly positive proteinuria, but no hematuria was detected by high-power microscopy. Blood gas analysis and biochemical tests indicated metabolic acidosis and electrolyte disorders, mainly manifested as hyponatremia, hypokalemia, hypocalcemia, hypophosphatemia, and hyperchloremia. The complication of Fanconi syndrome was considered. Further, we carried out humoral immunity test which revealed a decrease in complement C3 (0.38 g/L; the levels of C3 with the normal range was 0.70–2.06 g/L), and the complement C4 was normal (the normal range of C4 level was 0.11–0.61 g/L). Autoantibody ANA was positive, with the highest titer >1:3,200. Thyroid function was normal. Recent laboratory results were shown in Table 1. Urinary ultrasound indicated enhanced renal parenchymal echoes with unclear corticomedullary boundary, hydronephrosis in both kidneys, and no renal calcification or stones. The bone age was relatively below the standard, equivalent to 3.6-year-old or 10–25 percentiles. The contrast-enhanced voiding urosonography (VUS), CT urography (CTU), axial enhanced MRI of the sella turcica, conventional MRI of the lower limbs, and electromyography examination revealed no abnormalities. The whole-exome sequencing of the child did not found any suspected pathogenic gene mutations. After excluding diseases such as tuberculosis, hepatitis B, diabetes, genetic diseases and tumors, the patients were treated with sufficient oral methylprednisolone, captopril, potassium citrate, sodium citrate, potassium dihydrogen phosphate, and sodium dihydrogen phosphate for 28 days, but there was no improvement in proteinuria. Then renal biopsy was performed, and hematoxylin-eosin and other specific staining revealed 1/4 glomerular glomerulosclerosis. The remaining glomeruli showed mesangial cell and stroma mild hyperplasia, with thickening of the basement membrane and a small amount of spike like structures. Subepithelial deposition of immune complexes was observed. The staining also revealed granular and vacuolar degeneration of renal tubular epithelial cells, occasional protein tubular type, focal renal tubular lumen dilation accompanied by segmental epithelial cell detachment, brush border detachment, a few renal tubular atrophy, renal interstitial edema, small focal lymphoid and monocyte infiltration. There was no obvious lesion on the small artery wall. Immunofluorescence showed that IgG was deposited (++) in fine particles along the capillary loop. The electromicroscopic examination of ultrastructures showed mild irregular thickening of the basement membrane with a thickness of about 300–700 nm, diffused fusion of the foot processes, deposition of a large amount of electronic dense materials in the subepithelial and basement membranes, vacuolization and degeneration of the epithelial cells of the renal tubules, and no special changes in the renal interstitium. The pathological diagnosis was stage II membranous nephropathy with acute tubulointerstitial lesions. Paraffin section fluorescence staining showed IgG1 subtype+, IgG4++, while negative in IgG2, IgG3, PLA2R, and THSD7A staining, consistent with stage II membranous nephropathy (Figures 1–3). Clinical diagnosis of lupus nephritis (nephrotic syndrome type, V-type) was made, with a SLE disease activity index (SLEDAI) score of 14. After receiving a sufficient oral dose of methylprednisolone (12 mg, bid) for 60 days with the subsequent dosage tapered, and sequential anti-inflammatory treatment with prednisone acetate (10 mg, qd), immune suppression with mycophenolate mofetil (0.166 g, bid), 5 times of plasma exchange, captopril, and dipyridamole, the patient was discharged. A follow-up after taking the above oral drugs for 11 months showed that the symptoms of fatigue, excessive drinking, and polyuria improved, the complement C3 returned to normal levels, but there were still persistent proteinuria (2 + to 3+) and glycosuria (2 + to 4+); no hematuria was detected under high-power microscopy; SLEDAI score was 12; autoantibody ANA was positive with a titer of 1:320; and eGFR was calculated to be >30 ml/min/1.73 m2 (The eGFR at initial presentation was 53.1 ml/min/1.73 m2). Then the treatment with belimumab monoclonal antibody was added. After 9 times of regular use of belimumab, the clinical symptoms of the patient achieved complete remission; blood electrolytes, complement C3 and C4 returned to normal levels; and ANA titer was maintained at a relatively low level (1:320); urinary protein fluctuated between+to ∼+; urinary glucose fluctuated between+to 3+, the UTP was 0.785 g/24 h (24 h urine output of 1,700 ml), and eGFR was calculated to be 32.8 ml/min/1.73 m2, recent laboratory results were shown in Table 1. The dose of prednisone was reduced to 7.5 mg, qd. The follow-up examination showed a body-weight increase to the 50th percentile of the same age and gender, a height of the 3th percentile, and a SLEDAI score decrease to 4.
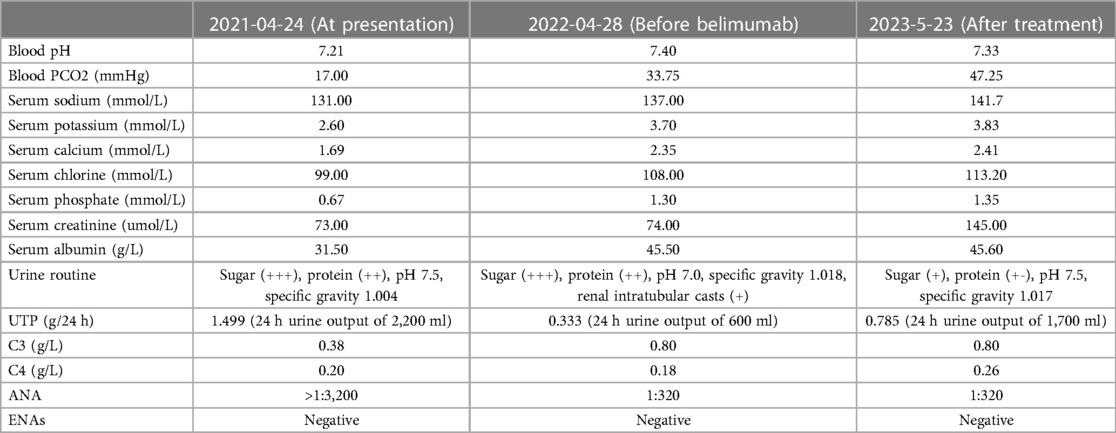
Table 1. Laboratory results.
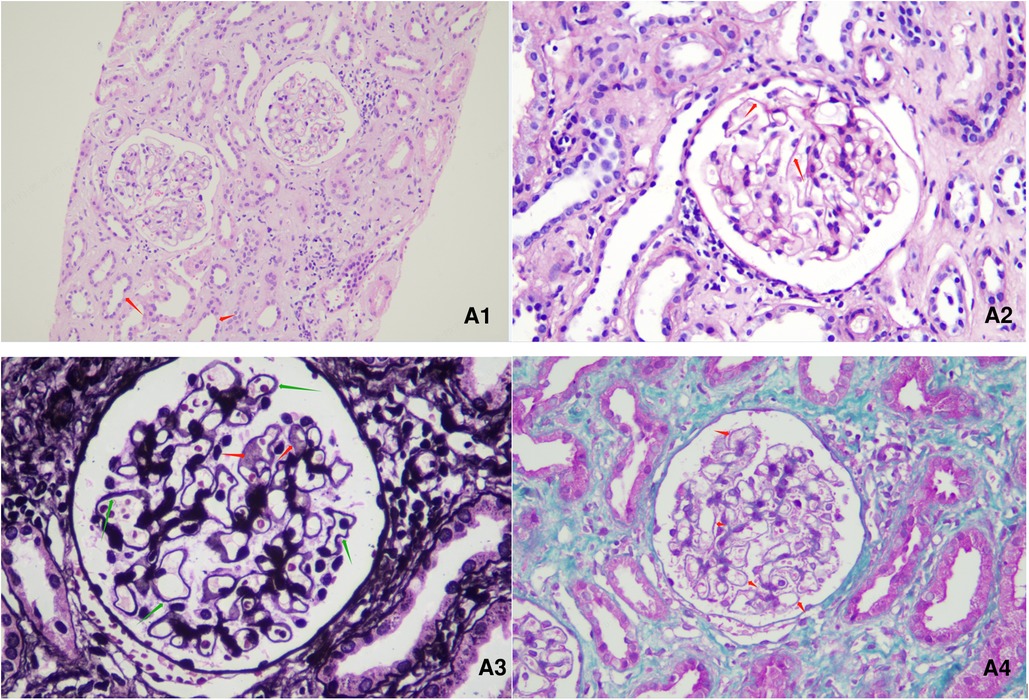
Figure 1. Renal pathological presentation of the patient. (A1): HE (hematoxylin-eosin) staining, ×200 magnification. (A2): PAS (periodic Acid Schiff), staining, ×400 magnification. (A3): PASM (periodic acid-silver methenamine) staining, ×600 magnification. (A4): Masson staining, ×400 magnification. Among the 4 glomeruli, glomerulosclerosis was observed in 1 glomerulus, while the other glomeruli showed glomerular mesangial cell and stroma mild hyperplasia. The capillary loops were open, with a stiff appearance. The basement membrane was thickened, with a small amount of spike-like structures visible. Subepithelial deposition of immune complexes was observed. No cellulose like necrosis, platinum ear-like structure or crescent formation was observed. The following changes were observed: granular and vacuolar degeneration of renal tubular epithelial cells, occasional protein tubular type, focal renal tubular lumen dilation accompanied by segmental epithelial cell detachment, brush border detachment, renal tubular atrophy, renal interstitial edema, small focal lymphoid and monocyte infiltration. There was no obvious lesion on the small artery wall.
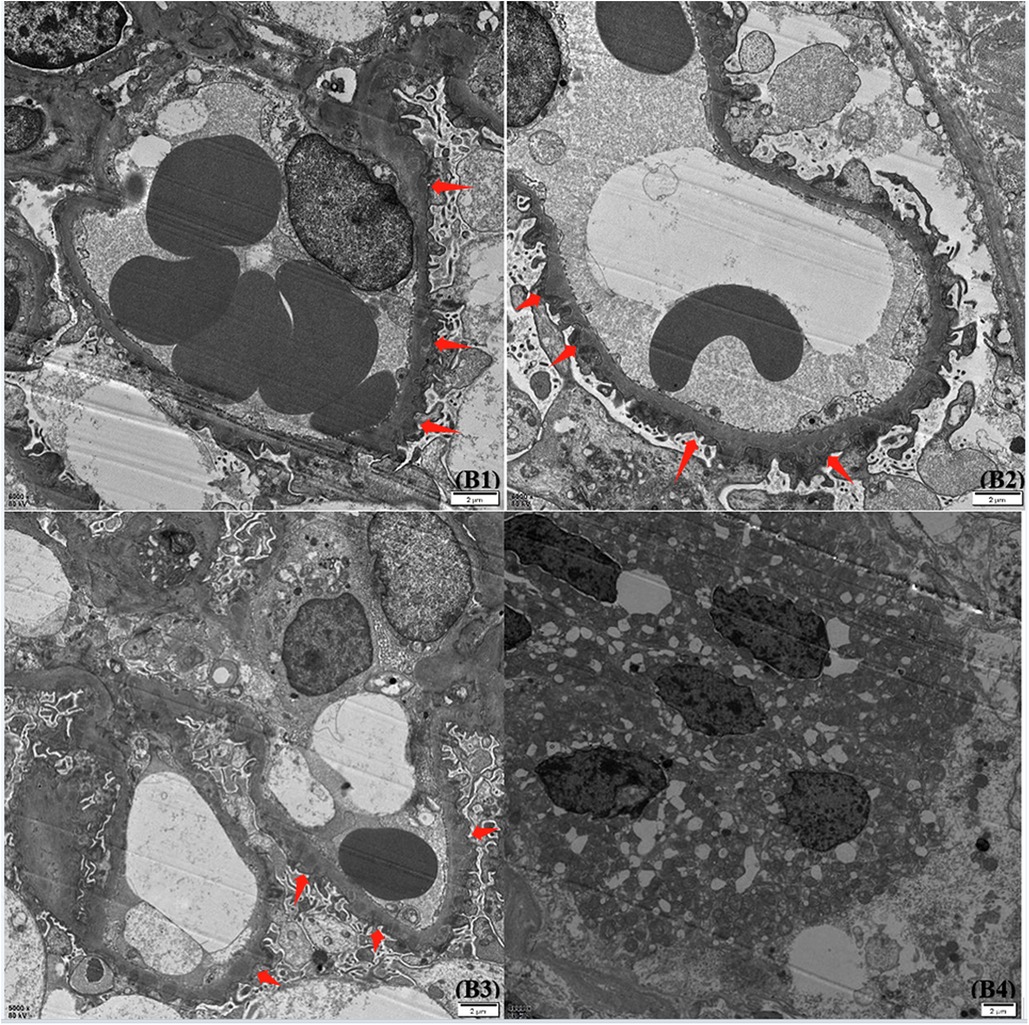
Figure 2. Glomerular findings by electron microscope. Under an electron microscope, the basement membrane showed mild irregular thickening, with a thickness of about 300–700 nm. The foot processes were diffusely fused, and a large amount of electronic dense material was deposited in the subepithelial and basement membranes. The epithelial cells of the renal tubules were vacuolated and degenerated, and there were no special changes in the renal interstitium.
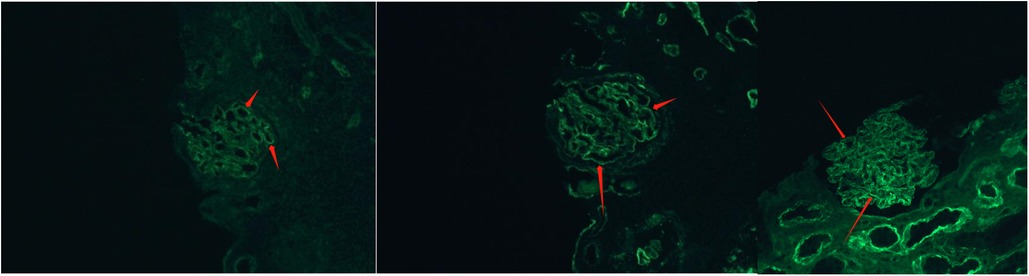
Figure 3. Glomerular findings by immunofluorescence staining. Epidemic immunofluorescence staining showed IgG deposits (++) in fine granular form along the capillary loop.
Discussion
SLE is a relatively rare systemic autoimmune disease. If not treated in a timely manner, irreversible damage to various organs can occur, ultimately leading to death (10). Childhood SLE refers to SLE that starts before the age of 18 years, accounting for 1/5 of all SLE cases (10). The onset age of cSLE in Asia is 8.6–13.5 years old, with only 5% of cases occurring before the age of 5 years (11). Compared with adult SLE, cSLE may have a higher correlation with genetic susceptibility, and cSLE has a more acute onset and more severe symptoms, with a higher probability of developing proteinuria, facial erythema, anti-dsDNA antibodies, hemolytic anemia, arthritis, etc. (12), Which further increasing the disability and mortality rate of the disease, lupus nephritis is one of the main causes of death in cSLE patients, 50%–70% of cSLE children can develop lupus nephritis (13–15), ranging from asymptomatic hematuria and/or proteinuria to rapidly progressive nephritis with renal function impairment, nephrotic syndrome, and end-stage kidney disease.
The clinical manifestations and initial symptoms of cSLE vary. About 60%–85% of children with SLE have atypical onset symptoms (3, 4). The onset age of the present case was young, and the symptoms were extremely atypical. The onset was characterized by fatigue, polydipsia, and polyuria. Combined with persistent metabolic acidosis and urinary pH >5.5, the patient was initially diagnosed with type I renal tubular acidosis (distal renal tubular acidosis, dRTA). Further complication was Fanconi syndrome based on the patient's normal blood glucose levels, sustained glycosuria, developmental delay, polyuria, thirst, hypophosphatemia, hypokalemia, metabolic acidosis, etc. Finally, the diagnosis of cSLE was made through renal biopsy. To our knowledge, there has been no report of cSLE combined with Fanconi syndrome. The case we reported was cSLE combined with type I renal tubular acidosis and Fanconi syndrome, which has not been previously reported.
SLE related kidney injuries may involve the glomerulus, tubulointerstitium, and vascular endothelium, but the involvement of the latter two have been rarely reported (16). Nowadays, the concept of SLE-associated tubulointerstitial nephritis (SLE TIN) is increasingly being proposed (17). Some scholars have pointed out that tubulointerstitial injury may be related to glomerular disease. At the same time, in the context of severe or active glomerular disease, interstitial changes in the tubules may not be obvious or even absent, or isolated tubular injury may occur in the absence of glomerular derived proteinuria (5, 18). The inconsistent occurrences suggest that there may be independent pathogenesis between tubular injury and glomerular disease (19). Tubulointerstitial injury is of great clinical significance, because it is not only a precursor to the occurrence of lupus, but also an independent predictive indicator of disease prognosis (20–22). SLE with renal tubular acidosis (RTA) has been widely reported. RTA is a kind of disease classified by clinical symptoms and biochemical features, which are characterized by the impairment of bicarbonate reabsorption or hydrogen secretion in different parts of renal tubules, leading to renal acidification dysfunction (23). The pathogenesis of SLE-related RTA is still unclear, which may be related to the deficiency of H+ -ATPase in intercalated cells on collecting duct caused by autoantibodies (18). The increase in membrane permeability of renal tubular lumen cells can also lead to the loss of a large amount of potassium ions along the concentration gradient, thereby inhibiting H+ -ATPase activity and leading to the disease development. Previous studies found that the presence of autoantibodies targeting carbonic anhydrase-II in patients with tubulointerstitial nephritis was associated with type-1 and type-3 RTA (15). Li et al. reported that 6 adult SLE patients were diagnosed with RTA in the later stage of SLE, with an average interval of 3 years between the two diagnoses; among the patients, 5 (83.3%) had proteinuria, and 1 (16.7%) had no significant proteinuria (14). Bagga and Nandi et al. reported that two children with RTA were initially diagnosed, but after targeted therapy, their conditions did not improve; the cause was repeatedly searched, and ultimately the patients were diagnosed with cSLE through renal tissue biopsy and strong serological evidence (24, 25). RTA may be the first symptom of SLE or may appear after diagnosis of SLE. RTA may be present from 1 month to 8 years after the onset of SLE, and fatigue and polyuria are common complaints in these children (14, 25). Renal biopsy of cSLE with RTA often show glomerular tissue hyperplasia, sclerosis, immune complex deposition, renal tubular atrophy, interstitial inflammation and fibrosis formation, consistent with our report. Among the abnormalities, tubulointerstitial lesions are considered as a strong predictor of renal function impairment and unfavourable long-term renal outcomes in LN (17, 21, 22, 26–29).
We searched the literature and found 7 case reports of cSLE combined with renal tubular acidosis, including 6 cases of cSLE combined with type 1 (distal type) renal tubular acidosis (24, 25, 30–33), and 1 case with unknown type of renal tubular acidosis (34). The basic clinical characteristics are shown in Table 2. These reports showed that SLE was often associated with type 1 renal tubular acidosis, with type 4 being rare, and type 2 (proximal type) even more rare (14, 17). In addition, previously reported cases of SLE combined with RTA were more common in other overlap syndromes, where these patients also had other immune diseases, such as autoimmune thyroiditis and Sjogren's syndrome (18, 35).
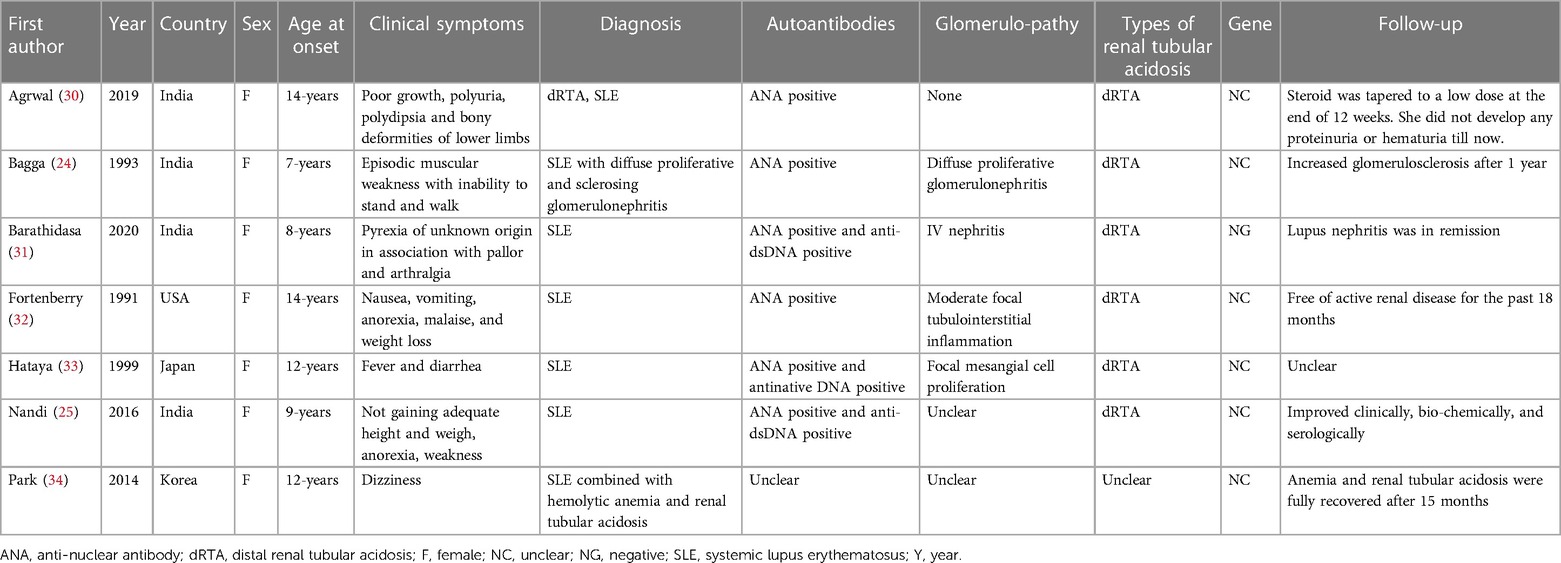
Table 2. Baseline characteristics of diagnosed of cSLE and dRTA.
Fanconi syndrome (FS), also known as Fanconi-Debré-de Toni syndrome or osteomalacia-renal glycosuria-aminoaciduria-hyperphosphateuria syndrome. FS can be congenital or acquired, transient or persistent, and can be accompanied by end-stage renal disease or a normal glomerular filtration rate (36–38). Previously, it was believed that the disease was hereditary, but in recent years, multiple clinical studies have suggested that its etiology is mostly acquired (39). Due to atypical symptoms and diverse causes, it is highly susceptible to misdiagnosis. Secondary FS is often secondary to immune diseases such as Sjogren's syndrome, multiple myeloma, anaphylactoid purpura, and can also be secondary to metabolic diseases such as galactose, hepatolenticular degeneration, or exposure to drugs and toxins that induce mitochondrial damage in kidneys (40, 41). cSLE combined with renal tubular acidosis often involves the distal renal tubules, yet the occurrence of Fanconi syndrome would indicate proximal renal tubular damage. This type of complication has not been previous reported. In our case, the renal pathology revealed stage II membranous nephropathy with acute tubulointerstitial injury. The pathogenic immune mechanism of SLE remains unclear, and the mechanism of proximal or distal tubular epithelial cell injury caused by lupus nephritis remains unclear. Some studies have shown that over-absorption of IgG anti-dsDNA antibodies and albumin by proximal tubular epithelial cells results in ROS-mediated tubular injury and interstitial inflammation (42). Fanconi syndrome may be caused by extensive proximal convoluted tubule injury caused by the disease. In our case, tubular injury was found in the kidney biopsy and renal intratubular casts were found in the urine routine, indicating this possibility. In addition, we suspected that most children with lupus nephritis do not have Fanconi syndrome, which may be related to the strong compensatory capacity of the glomeruli in children and the lack of serious renal interstitial damage. The severity of proximal renal tubule injury may be the key condition for the occurrence of typical Fanconi syndrome, but the specific mechanism needs further study.
The case reported here was prone to misdiagnosis in clinical practice, since the age of the child was not within the typical age range for childhood SLE, and the clinical symptoms were extremely atypical. If the renal biopsy findings were unavailable, the clinical manifestations would not meet the diagnostic criteria for cSLE, and the symptoms of the child such as fatigue significantly improved after the treatment for renal tubular acidosis, making it easy to misdiagnose. This patient had refractory hyperchloremic metabolic acidosis, hypokalemia, alkaline urine (urine pH >5.5), and persistent proteinuria, as well as manifestations of renal glycosuria, proteinuria, refractory electrolyte disorders, growth and development retardation combined with Fanconi syndrome. Before treatment, we conducted detailed medical history inquiry, physical examination and examination of the child. There were no patients with long-term chronic kidney disease in his family, and the simple examination of urine and kidney function of the parents of the child showed no abnormalities. In addition, the whole exon sequencing test showed no suspected pathogenic gene variation, and the regular child health examination in the local hospital before the child was 3 years old showed no obvious abnormalities, so the possibility of FS caused by congenital or genetic factors were not supported. Acquired FS usually occurs secondary to diseases or exposure to certain toxins or drugs, but a detailed history did not reveal the consumption of any potentially nephrotoxic medications or exposure to radiation or environmental toxin. Furthermore, the patient was not complicated with Henoch–Schonlein purpura, liver disease, diabetes, thyroid disease, chronic infection, tumor, or metabolic disease, and there was no strong evidence to support obstructive nephropathy, so it was highly suspected that cSLE lesions involved renal tubules combined with acquired FS.
However, it was questionable why the child has significant growth delay and a decrease in eGFR at the time of presentation. On the one hand, we believed that the symptoms of SLE in children were too insidious, resulting in family members not finding abnormalities in time, and the child was too young to accurately explain his physical discomfort. On the other hand, kidney damage caused by the disease may be compensated by a subset of normal nephrons, so symptoms were masked. Based on the above two aspects, it is speculated that the child may have a longer course of disease than the chief complaint, so there has been significant growth retardation and decline in kidney function at the time of presentation. The above conclusions further indicate that atypical cSLE is difficult to detect due to insidious symptoms, and delayed medical treatment may cause irreversible damage to kidney function, so doctors need to be more sensitive to find the problem and diagnose it in time.
LN is by default severe disease, that can lead to gradual nephron loss and chronic kidney disease (CKD) in addition to death. belimumab is currently the only biological agent approved for the treatment of cSLE, achieving therapeutic effect primarily by binding to B-lymphocyte activating factors (43). In a post-hoc analysis of the BLISS-LN, belimumab was found to reduce the risk for flares by 55% compared with standard-of-care (SoC, low-dose CYC62 or mycophenolate in combination with GC) alone, and preserve glomerular filtration rate (GFR) better than SoC (44). Belimumab may be considered as an add-on therapy in patients with refractory or relapsed SLE to reduce disease activity, disease recurrence rate, and hormonal dose. But the current recommendations do not require prior failure to one or more conventional drugs before initiating a biological agent. It is worth noticing that after nearly a year of combined treatment with immunosuppressive agents and corticosteroids, the patient's high levels of proteinuria and glycosuria were not corrected satisfactorily. Considering that the pathological type of the patient's lupus nephritis was nephrotic syndrome type (V-type) with renal tubulointerstitial injury, a case of refractory lupus nephritis. After fully communicating with the child's family and obtaining their consent, we added the biological agent belimumab into the therapy. The patient's clinical symptoms, such as fatigue, were completely relieved after 9 times of regular use of belimumab. In addition, the patient's blood electrolytes, complement C3 and C4 returned to normal levels; ANA titer decreased to 1:320; urinary protein fluctuated between urinary glucose fluctuated between + to ∼+; urinary glucose fluctuated between + to 3 + . The dose of prednisone was smoothly tapered to 7.5 mg, qd. The patient's body-weight increased to the 50th percentile of the same age and gender; his height was in the 3th percentile; and SLEDAI score decreased to 4 on the most recent follow-up. Currently, his 24-hour urinary protein is close to a normal level. These results suggest that belimumab is effective in the treatment of the patient's disease.
In conclusion, the clinical manifestations of SLE in children can be very insidious, and tubular acidosis may be the first and prominent manifestation of childhood lupus nephritis. So far, there has been no report of secondary Fanconi syndrome in children with lupus nephritis. Therefore, for children with symptoms such as unexplained renal tubular acidosis, hypokalemia, proteinuria, and renal glycosuria, it is necessary to be alert to atypical cSLE in addition to congenital and genetic metabolic diseases. It is recommended to carry out long-time follow-ups, screen for autoantibodies and perform other immune tests in a timely manner. When a clinical diagnosis is difficult, a clear diagnosis can be made through renal puncture biopsy, so that the patient can be diagnosed and treated early with an improved prognosis.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
LL contributed to conceptualization, writing of original draft, reviewing and editing. HG and MS contributed to conceptualization, reviewing and editing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the grants from the Science and Technology Bureau of Sichuan province (No. 21ZDYF1329).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kamphuis S, Silverman ED. Prevalence and burden of pediatric-onset systemic lupus erythematosus. Nat Rev Rheumatol. (2010) 6(9):538–46. doi: 10.1038/nrrheum.2010.121
2. Harry O, Yasin S, Brunner H. Childhood-onset systemic lupus erythematosus: a review and update. J Pediatr. (2018) 196:22–30.e2. doi: 10.1016/j.jpeds.2018.01.045
3. Bader-Meunier B, Armengaud JB, Haddad E, Salomon R, Deschênes G, Koné-Paut I, et al. Initial presentation of childhood-onset systemic lupus erythematosus: a French multicenter study. J Pediatr. (2005) 146(5):648–53. doi: 10.1016/j.jpeds.2004.12.045
4. Iqbal S, Sher MR, Good RA, Cawkwell GD. Diversity in presenting manifestations of systemic lupus erythematosus in children. J Pediatr. (1999) 135(4):500–5. doi: 10.1016/S0022-3476(99)70174-5
5. Levy DM, Kamphuis S. Systemic lupus erythematosus in children and adolescents. Pediatr Clin North Am. (2012) 59(2):345–64. doi: 10.1016/j.pcl.2012.03.007
6. Kawasaki Y, Ohara S, Miyazaki K, Kanno S, Ono A, Suyama K, et al. Incidence and prognosis of systemic lupus erythematosus in a 35 year period in Fukushima, Japan. Pediatr Int. (2015) 57(4):650–5. doi: 10.1111/ped.12588
7. Wu CY, Li CF, Wu QJ, Xu JH, Jiang LD, Gong L, et al. Chinese systemic lupus erythematosus treatment and research group registry Ⅸ:clinical features and survival of childhood onset systemic lupus erythematosus in China. Chin Med J (Engl). (2017) 130(11):1276–82. doi: 10.4103/0366-6999.206346
8. Hiraki LT, Benseler SM, Tyrrell PN, Hebert D, Harvey E, Silverman ED. Clinical and laboratory characteristics and long-term outcome of pediatric systemic lupus erythematosus: a longitudinal study. J Pediatr. (2008) 152(4):550–6. doi: 10.1016/j.jpeds.2007.09.019
9. Dang XQ, Yi ZW. Interpretation of evidence-based guideline on diagnosis and treatment of lupus nephritis (2016). Chinese Journal of Pediatrics. (2018) 2:95–9. doi: 10.3760/cma.j.issn.0578-1310.2018.02.004
10. Tsokos GC. Autoimmunity and organ damage in systemic lupus erythematosus. Nat Immunol. (2020) 21:605–14. doi: 10.1038/s41590-020-0677-6
11. Aggarwal A, Srivastava P. Childhood onset systemic lupus erythematosus: how is it different from adult SLE? Int J Rheum Dis. (2015) 18(2):182–91. doi: 10.1111/1756-185X.12419
12. Webb R, Kelly JA, Somers EC, Hughes T, Kaufman KM, Sanchez E, et al. Early disease onset is predicted by a higher genetic risk for lupus and is associated with a more severe phenotype in lupus patients. Ann Rheum Dis. (2011) 70(1):151–6. doi: 10.1136/ard.2010.141697
13. Li SL, Liou LB, Fang JT, Tsai WP. Symptomatic renal tubular acidosis (RTA) in patients with systemic lupus erythematosus: an analysis of six cases with new association of type 4 RTA. Rheumatology (Oxford). (2005) 44(9):1176–80. doi: 10.1093/rheumatology/keh705
14. Inagaki Y, Jinno-Yoshida Y, Hamasaki Y, Ueki H. A novel autoantibody reactive with carbonic anhydrase in sera from patients with systemic lupus erythematosus and sjögren’s syndrome. J Dermatol Sci. (1991) 2(3):147–54. doi: 10.1016/0923-1811(91)90060-B
15. Fanconi G. Die nicht diabetischen glykosurien und hyperglykaemien des aelteren kindes. Jahrbuch Fuer Kinderheilkunde. (1931) 133:257–300.
16. Rawla P, Thandra KC, Aluru JS, Mageed SA, Sakr EE, Elsayed GG, et al. Systematic review and case report: systemic lupus erythematosus with renal tubular acidosis. Clin Case Rep. (2020) 8(2):333–40. doi: 10.1002/ccr3.2623
17. Dhingra S, et al. Tubulointerstitial nephritis in systemic lupus erythematosus: innocent bystander or ominous presage. Histol Histopathol. (2014) 29(5):553–65. doi: 10.14670/HH-29.10.553
18. Kozeny GA, Barr W, Bansal VK, Vertuno LL, Fresco R, Robinson J, et al. Occurrence of renal tubular dysfunction in lupus nephritis. Arch Intern Med. (1987) 147(5):891–5. doi: 10.1001/archinte.1987.00370050087015
19. D’Agati V. Renal disease in systemic lupus erythematosus, mixed connective tissue disease, sjogren’s syndrome, and rheumatoid arthritis. In: Jennette JC, Olson JL, Schwartz MM, Silva FG, editors. Heptinstall’s Pathology of Kidney. 6th edn. Philadelphia: Lippincott-Raven (2007). p. 517–612.
20. Howie AJ, Turhan N, Adu D. Powerful morphometric indicator of prognosis in lupus nephritis. QJM. (2003) 96(6):411–20. doi: 10.1093/qjmed/hcg074
21. Yu F, Wu LH, Tan Y, Li LH, Wang CL, Wang WK, et al. Tubulointerstitial lesions of patients with lupus nephritis classified by the 2003 international society of nephrology and renal pathology society system. Kidney Int. (2010) 77(9):820–9. doi: 10.1038/ki.2010.13
22. Hsieh C, Chang A, Brandt D, Guttikonda R, Utset TO, Clark MR. Predicting outcomes of lupus nephritis with tubulointerstitial inflammation and scarring. Arthritis Care Res (Hoboken). (2011) 63(6):865–74. doi: 10.1002/acr.20441
23. Morris RC Jr. Renal tubular acidosis. Mechanisms, classification and implications. N Engl J Med. (1969) 281(25):1405–13. doi: 10.1056/NEJM196912182812508
24. Bagga A, Jain Y, Srivastava RN, Bhuyan UN. Renal tubular acidosis preceding systemic lupus erythematosus. Pediatr Nephrol. (1993) 7(6):735–6. doi: 10.1007/BF01213337
25. Nandi M, Das MK, Nandi S. Failure to thrive and nephrocalcinosis due to distal renal tubular acidosis: a rare presentation of pediatric lupus nephritis. Saudi J Kidney Dis Transpl. (2016) 27(6):1239–41. doi: 10.4103/1319-2442.194679
26. Rijnink EC, Teng YKO, Wilhelmus S, Almekinders M, Wolterbeek R, Cransberg K, et al. Clinical and histopathologic characteristics associated with renal outcomes in lupus nephritis. Clin J Am Soc Nephrol. (2017) 12(5):734–43. doi: 10.2215/CJN.10601016
27. Hill GS, Delahousse M, Nochy D, Mandet C, Bariéty J. Proteinuria and tubulointerstitial lesions in lupus nephritis. Kidney Int. (2001) 60(5):1893–903. doi: 10.1046/j.1523-1755.2001.00017.x
28. Daniel L, Sichez H, Giorgi R, Dussol B, Figarella-Branger D, Pellissier JF, et al.. Tubular lesions and tubular cell adhesion molecules for the prognosis of lupus nephritis. Kidney Int. (2001) 60(6):2215–21. doi: 10.1046/j.1523-1755.2001.00055.x
29. Weening JJ, D'Agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int. (2004) 65(2):521–30. doi: 10.1111/j.1523-1755.2004.00443.x
30. Agrwal S, Mantan M, Dabas A. An unusual case of familial systemic lupus erythematosus with distal renal tubular acidosis and hemolytic anemia. Iran J Kidney Dis. (2019) 13(5):337–9.31705751
31. Barathidasan GS, Krishnamurthy S, Karunakar P, Rajendran R, Ramya K, Dhandapany G, et al. Systemic lupus erythematosus complicated by a gitelman-like syndrome in an 8-year-old girl. CEN Case Rep. (2020) 9(2):129–32. doi: 10.1007/s13730-019-00440-1
32. Fortenberry JD, Kenney RD. Distal renal tubular acidosis as the initial manifestation of systemic lupus erythematosus in an adolescent. J Adolesc Health. (1991) 12(2):148–51. doi: 10.1016/0197-0070(91)90458-X
33. Hataya H, Ikeda M, Ide Y, Kobayashi Y, Kuramochi S, Awazu M. Distal tubular dysfunction in lupus nephritis of childhood and adolescence. Pediatr Nephrol. (1999) 13(9):846–9. doi: 10.1007/s004670050713
34. Park JH, Im JW, Jun HK, Park HM, Choi SW, Park SK, et al. Delayed and long-term remission of refractory hemolytic Anemia in a child with systemic lupus erythematosus treated with rituximab. J Rheum Dis. (2014) 21(4):196–200. doi: 10.4078/jrd.2014.21.4.196
35. Fang JT, Chen YC. Systemic lupus erythematosus presenting initially as hydrogen ATPase pump defects of distal renal tubular acidosis. Ren Fail. (2000) 22(5):647–52. doi: 10.1081/JDI-100100906
36. Kleta R, Blair SC, Bernardini I, Kaiser-Kupfer MI, Gahl WA. Keratopathy of multiple myeloma masquerading as corneal crystals of ocular cystinosis. Mayo Clin Proc. (2004) 79(3):410–2. doi: 10.4065/79.3.410
37. Magnano L, Fernández de Larrea C, Cibeira MT, Rozman M, Tovar N, Rovira M, et al. Acquired fanconi syndrome secondary to monoclonal gammopathies: a case series from a single center. Clin Lymphoma Myeloma Leuk. (2013) 13(5):614–8. doi: 10.1016/j.clml.2013.04.008
38. Sirac C, Bridoux F, Essig M, Devuyst O, Touchard G, Cogné M. Toward understanding renal fanconi syndrome: step by step advances through experimental models. Contrib Nephrol. (2011) 169:247–61. doi: 10.1159/000313962
39. Zheng F, Zhao S, Li X. Clinical characteristics and biochemical abnormalities of fanconi syndrome. Chinese Journal of Internal Medicine. (2000) 11:14–7.
40. Chen Z, Zhang L, Chen L. The mechanism of energy metabolism disorders in proximal renal tubules leading to fanconi syndrome. Pediatr Res. (2019) 7:544–7. doi: 10.3760/cma.j.issn.1001-7097.2019.07.012
41. Heidari R. The footprints of mitochondrial impairment and cellular energy crisis in the pathogenesis of xenobiotics-induced nephrotoxicity, serum electrolytes imbalance, and fanconi’s syndrome: a comprehensive review. Toxicology. (2019) 423:1–31. doi: 10.1016/j.tox.2019.05.002
42. Alli AA, Desai D, Elshika A, Conrad M, Proneth B, Clapp W, et al. Kidney tubular epithelial cell ferroptosis links glomerular injury to tubulointerstitial pathology in lupus nephritis. Clin Immunol. (2023) 248:109213. doi: 10.1016/j.clim.2022.109213
43. Klavdianou K, Lazarini A, Fanouriakis A. Targeted biologic therapy for systemic lupus erythematosus: emerging pathways and drug pipeline. BioDrugs. (2020) 34(2):133–47. doi: 10.1007/s40259-020-00405-2
Keywords: atypical, childhood systemic lupus erythematosus, lupus nephritis, renal tubular acidosis, Fanconi syndrome
Citation: Lou L, Guo H and Shao M (2024) Systemic lupus erythematosus complicated with Fanconi syndrome: a case report and literature review. Front. Pediatr. 11:1230366. doi: 10.3389/fped.2023.1230366
Received: 28 May 2023; Accepted: 14 December 2023;
Published: 5 January 2024.
Edited by:
Orkun Tolunay, University of Health Sciences, TürkiyeReviewed by:
Jerome Lane, Ann & Robert H. Lurie Children’s Hospital of Chicago, United StatesSwee Ping Tang, Hospital Selayang, Malaysia
Sern Chin Lim, MARA University of Technology, Malaysia
© 2024 Lou, Guo and Shao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Guo Z3VvaHVpMDMyOUAxNjMuY29t Meiying Shao c2hhb21laXlpbmcyMDEzQDE2My5jb20=