 Sanya Thomas1,2*
Sanya Thomas1,2* Geoffrey Guenther2,3Jared H. Rowe2,4
Geoffrey Guenther2,3Jared H. Rowe2,4 Craig D. Platt2,5
Craig D. Platt2,5 Akiko Shimamura2,6
Akiko Shimamura2,6 Ofer Levy1,2,3,7Lakshmi Ganapathi2,8,9
Ofer Levy1,2,3,7Lakshmi Ganapathi2,8,9
- 1Precision Vaccines Program, Department of Pediatrics, Boston Children’s Hospital, Boston, MA, United States
- 2Harvard Medical School, Boston, MA, United States
- 3Division of Infectious Diseases, Boston Children’s Hospital, Boston, MA, United States
- 4Division of Hematology, Boston Children’s Hospital and Division of Pediatric Oncology, Dana-Farber Cancer Institute, Boston, MA, United States
- 5Division of Immunology, Boston Children’s Hospital, Boston, MA, United States
- 6Division of Hematology/Oncology, Boston Children's Hospital and Department of Pediatric Oncology, Dana-Farber Cancer Institute, Boston, MA, United States
- 7Broad Institute of MIT & Harvard, Cambridge, MA, United States
- 8Division of Pediatric Global Health, Massachusetts General Hospital, Boston, MA, United States
- 9Division of Pediatric Infectious Diseases, Massachusetts General Hospital, Boston, MA, United States
Severe congenital neutropenia caused by jagunal homolog 1 (JAGN1) mutation is a rare condition resulting from maturation arrest secondary to endoplasmic reticulum stress response from impaired neutrophil protein glycosylation. Here, we report a case of a 4-year-old boy who presented with a history of recurrent infections and manifestations, including recurrent intracranial hemorrhage. A review of similar cases reported in the literature indicates that a bleeding diathesis has not been previously described in these patients. We hypothesize that this newly described association of bleeding complications in this patient with JAGN1 mutation is secondary to defective glycosylation in the normal functioning of platelets or clotting factors. Recurrent infections with intracranial hemorrhage, new focal neurologic defects, or altered mental status in a child should warrant a suspicion for this immunodeficiency for the prompt initiation of treatment and prophylaxis for life-threatening infections or trauma.
Background
Neutrophils are key effectors of innate immunity that contribute to defense against bacterial and fungal infections (1). Neutropenia is usually acquired, resulting from increased destruction, granulocyte apoptosis, or decreased granulocyte production. Severe congenital neutropenia (SCN) is a rare primary immunodeficiency resulting in recurrent infections (2). Patients with congenital neutropenia are at an increased risk of acquiring myelodysplastic syndrome or acute myeloblastic leukemia (3). Jagunal homolog 1 (JAGN1) is a protein important for neutrophil maturation and survival (4). Mutations in the gene encoding JAGN1 are a rare and recently identified cause of SCN characterized by recurrent infections along with bony abnormalities and a heterogeneous clinical presentation (3).
Here, we report a case of a patient with autosomal recessive JAGN1 deficiency leading to SCN, with recurrent infections associated with bleeding manifestations. To our knowledge, bleeding manifestations associated with JAGN1 deficiency have not been previously reported, and this may be the first case of JAGN1 neutropenia related to biallelic pathogenic heterozygous mutations. We also review the literature for other cases of congenital neutropenia resulting from JAGN1 deficiency.
Case presentation
A 4-year-old boy presented with high-grade fever and recurrent right periorbital swelling following the recent treatment of right periorbital cellulitis and right ethmoidal sinusitis with broad-spectrum antibiotics for 2 weeks. His medical history was notable for the following: at birth, bilateral temporoparietal intraparenchymal hemorrhagic infarcts were noted with associated residual facial weakness. An extensive evaluation did not reveal any bleeding disorder or any underlying anatomic cause for his intracranial hemorrhage. He had an absolute neutrophil count (ANC) of 40 cells/µl, initially ascribed to acute illness. At 2 weeks of age, he developed a spontaneously draining abscess in his right groin. Abscess drainage culture revealed methicillin-resistant Staphylococcus aureus (MRSA). At that time, his ANC was 10 cells/µl. Bone marrow aspirate demonstrated myeloid maturation arrest with a few mature myeloid forms. Erythroid elements were of limited quantity and exhibited full-spectrum maturation. Megakaryocytes exhibited normal morphology. Genetic analysis was performed using targeted capture for genes associated with SCN (the University of Washington Department of Laboratory Medicine, MarrowSeq Panel, which includes ABCB7, ADA, AK2, ANKRD26, ATM, ATR, ATRX, BLM, BRCA1, BRCA2, BRIP1, C150RF41, CBL, CDAN1, CEBPA, CSF3R, CTC1, CXCR4, DKC1, ELANE, ERCC4, ETV6, FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FANCM, G6PC3, GATA1, GATA2, GFI1, HAX1, IL2RG, JAGN1, JAK2, KIF23, KLF1, LIG4, LYST, MPL, MRE11A, NBN, NHP2, NOP10, PALB2, PAX5, RAB27A, RAD50, RAD51C, RBM8A, RMRP, RNF168, RPL10, RPL11, RPL26, RPL35A, RPL5, RPS10, RPS14, RPS17, RPS19, RPS24, RPS26, RPS7, RTEL1, RUNX1, SBDS, SEC23B, SLX4, SRP72, TAZ, TCIRG1, TERC, TERT, TINF2, TP53, USB1, VPS45, WAS, and WRAP53), followed by next-generation sequencing with Illumina technology, which revealed biallelic pathogenic heterozygous mutations (p.S64X, NM_032492.3:c.191C > G and p.Q127X, NM_03492.3:c.379C > T) in the JAGN1 gene.
The patient was treated with granulocyte colony-stimulating factor (GCSF). However, his response to GCSF was inconsistent even with higher doses of 10 mcg/kg/day, with widely fluctuating ANC values (Figure 1). Additional screening revealed hypogammaglobulinemia with an IgG level of 53 mg/dl at 5 months of age (normal range: 200–1,200 mg/dl). Therefore, he was started on immunoglobulin replacement therapy as well. Despite these interventions, he continued to develop recurrent infections, including skin and soft tissue infections as well as chronic nasal congestion. At 1 year of age, after sustaining minor head trauma following a fall, he developed a venous hemorrhagic cerebral infarct and a retro-clival and cerebellopontine angle cistern hematoma. At 2 years of age, he experienced acute respiratory failure and bradycardic cardiac arrest attributed to an aspiration event while receiving non-invasive positive pressure ventilation after undergoing adenotonsillectomy for obstructive sleep apnea. Until the performance of the tonsillectomy, he was on continuous positive airway pressure (CPAP) for obstructive sleep apnea. In addition, he developed right parasagittal watershed infarcts between the anterior and the middle cerebral arteries with subsequent left hemiparesis. Several months later, he experienced a focal seizure and Todd's paralysis and was treated with levetiracetam for seizure prevention.
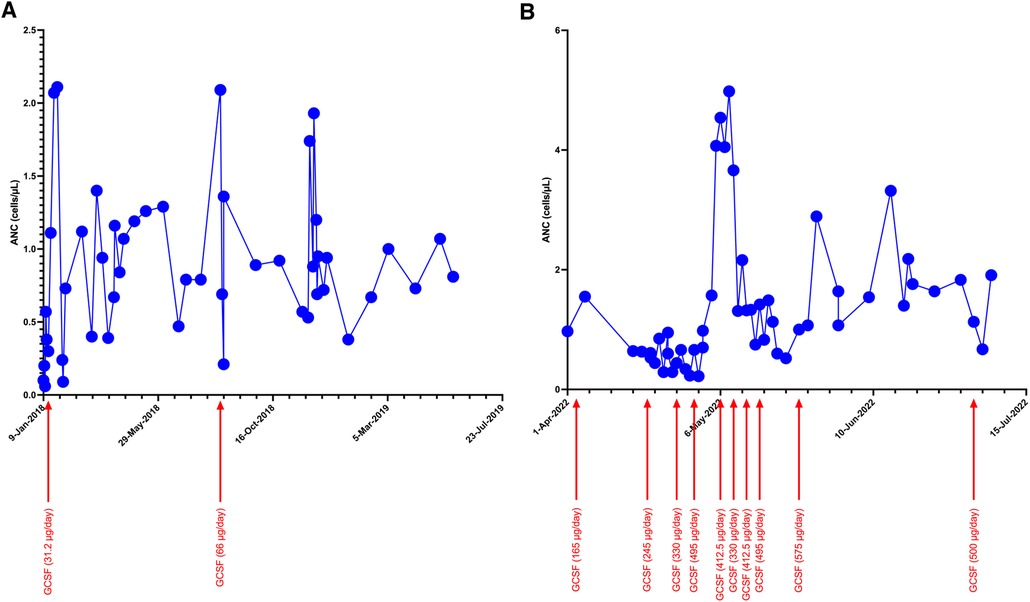
Figure 1. Trends of ANC during initial (A) and current (B) hospitalization with increasing doses of GCSF.
On presentation, he was alert, febrile, tachycardic, and tachypneic. He had mild right periorbital edema and erythema. Neurologic examination was significant for mild left lower facial weakness and mild left upper-extremity weakness, with slightly exaggerated reflexes on the left side globally. No dysmorphic features were observed. His height-for-age was under the 5th percentile, and his weight-for-age was between the 15th and the 25th percentile. A partially treated bacterial infection following his recent hospitalization was suspected. A brain computed tomography (CT) revealed complicated acute sinusitis with an epidural abscess. Magnetic resonance imaging (MRI) confirmed an epidural abscess with adjacent cerebritis, which was thought to be reactive in nature (Figure 2). He was hospitalized for further care.
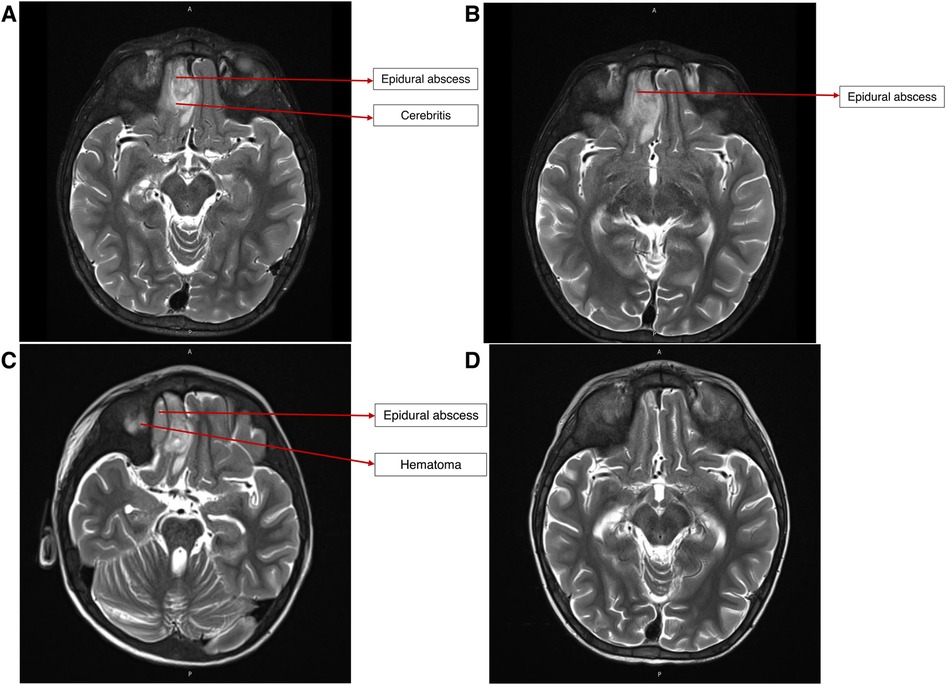
Figure 2. (A) MRI brain on presentation, demonstrating epidural abscess and reactive cerebritis. (B) MRI brain after endoscopic drainage procedure, showing persistence of epidural abscess and cerebritis. (C) MRI brain after craniotomy and debridement, showing persistence of epidural abscess and new adjacent hematoma. (D) MRI brain after ∼6 weeks of broad-spectrum antibiotic therapy, showing no abscess or residual parenchymal changes.
During hospitalization, hematologic studies revealed anemia, a normal leukocyte count with neutropenia and monocytosis, thrombocytopenia, and elevated C-reactive protein (CRP) (Table 1). Initial blood cultures, cerebrospinal fluid (CSF) analysis, and CSF culture did not reveal anything significant. He was treated empirically with vancomycin, metronidazole, and cefepime. He underwent endoscopic drainage of the abscess, which demonstrated growth of Pseudomonas aeruginosa on culture, which was sensitive to cefepime. Nevertheless, a broad antibiotic coverage was maintained, given the inability to exclude polymicrobial infection in the context of recent antibiotic use. Achieving therapeutic vancomycin trough levels was challenging, prompting a change to daptomycin. An interval imaging demonstrated an increased size of the abscess after endoscopic surgery, and he subsequently underwent frontal craniotomy.

Table 1. Investigations on presentation.
After craniotomy, an interval MRI demonstrated a new epidural collection lateral to the epidural abscess, which was thought to represent a postoperative hematoma. A final interval MRI obtained 5 weeks after craniotomy showed a resolution of the abscess and hematoma.
Because of the nature of his surgeries, an alternative to CPAP was preferred, ultimately leading to the performance of a tracheostomy. Early during his course of stay, ANC dropped to 0, and GCSF dosing increased to ∼40 μg/kg/day to maintain an ANC >1,000 cells/µl. Because of his bleeding history, sequential compression device boots were employed while he was immobile, instead of using systemic anticoagulation.
During his hospital stay, the patient developed a febrile illness, and a blood culture taken from his central venous catheter grew Candida lusitaniae (Clavispora lusitaniae). Fungal staging examinations showed no evidence of fungal infection in CSF culture, ocular exam, or abdominal ultrasonography. His central venous catheter was removed, and he was treated with micafungin for 2 weeks.
Shortly after the patient completed a 6-week course of cefepime, daptomycin, and metronidazole for epidural abscess, his hospital course was complicated by a new requirement for supplemental oxygen and by a new retrocardiac opacity on chest x-ray, and he was diagnosed with pneumonia, which was treated with cefepime for 7 days.
His hospital stay is summarized in Table 2. On the day of discharge, his ANC was 1,910 cells/µl, and he was hemodynamically stable.
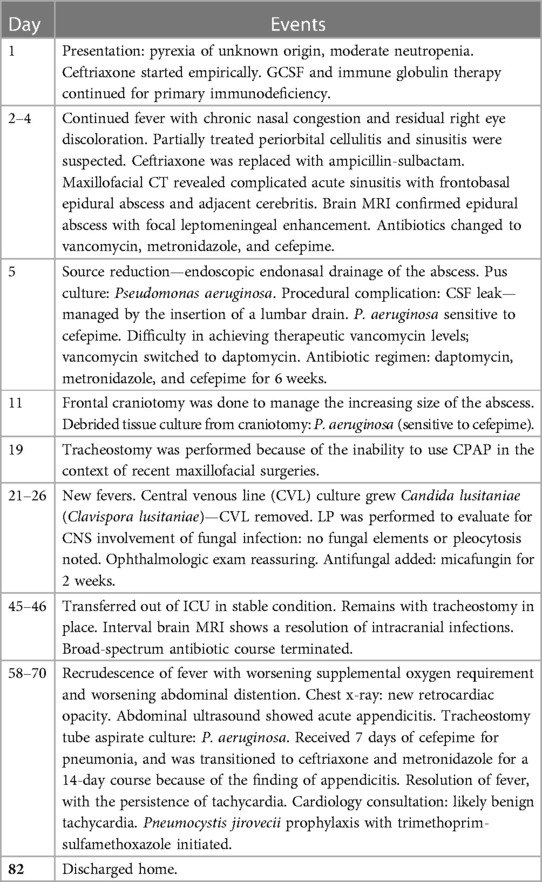
Table 2. Course during the hospital stay.
He was readmitted approximately 2 months after discharge for planned sibling donor allogeneic hematopoietic stem cell transplantation.
Discussion
Herein, we describe a rare case of a patient with SCN associated with biallelic JAGN1 mutations. SCN is uncommon, with associated genetic mutations occurring in >20 genes, including ELANE, GFI1, HAX1, G6PC3, WAS, and VPS45, and diverse inheritance patterns that can be autosomal-dominant, autosomal-recessive, or X-linked (5, 6). SCN caused by JAGN1 deficiency is extremely rare, accounting for ∼10% of cases (5).
JAGN1 is expressed in the endoplasmic reticulum (ER), which contributes to the early secretory pathway in the ER and is a critical regulator of neutrophil differentiation and survival via GCSF receptor-mediated signaling (4). Biallelic mutations in JAGN1 lead to several defects in the granulocyte structure and cellular function and also affect longevity (4). A knockdown of JAGN1 expression in HeLa cells interferes with STAT3 phosphorylation upon recombinant human GCSF treatment, suggesting that decreased GCSF receptor signaling may contribute to defective granulocytes (4). Granulocyte-macrophage colony–stimulating factor (GM-CSF) treatment of bone marrow granulocytes in patients with JAGN1 deficiency restores the phosphorylation of STAT5 and cytotoxicity in response to Candida albicans (7). JAGN1 deficiency is also associated with a decreased expression of myeloperoxidase (MPO) in neutrophils, contributing to an ineffective killing of C. albicans via neutrophil extracellular traps—a phenotype reversible via GM-CSF administration (8). Mutations in JAGN1 can result in ER stress and intracellular calcium activation of calpain, leading to myeloid cell apoptosis (Figure 3) (9).
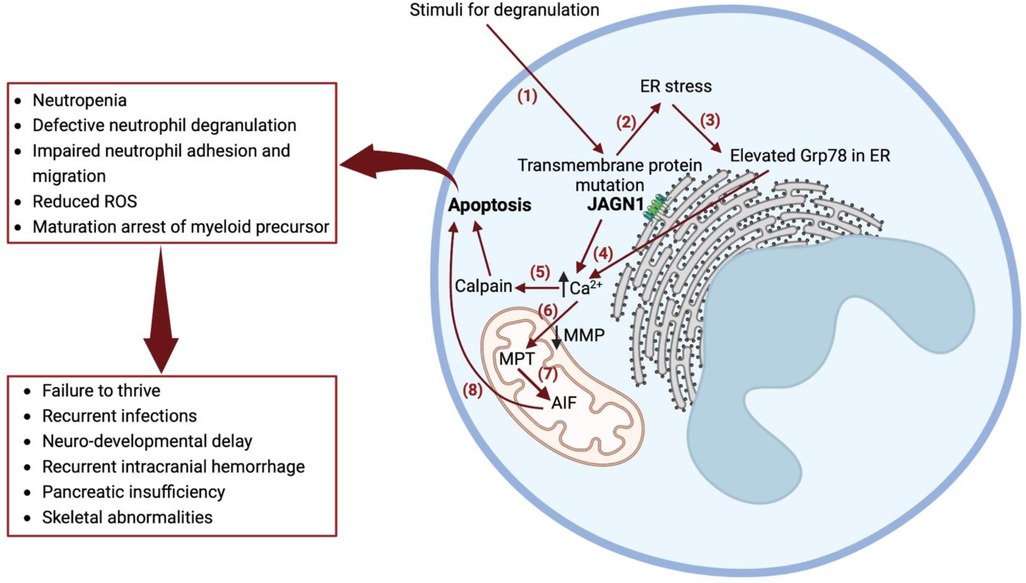
Figure 3. Mechanism of severe congenital neutropenia caused by JAGN1 deficiency. (1) Stimuli for degranulation in a wild-type JAGN1 transmembrane protein produces degranulation. (2) Mutant JAGN1 produces ER stress. (3) ER stress results in increased Grp78 protein. (4) Elevated Grp78 causes N-glycosylation. (5) Altered N-glycosylation results in elevated calcium in the cytoplasm, causing activation of calpain, which leads to apoptosis. (6) Reduced MMP leads to Ca2+ entry to mitochondria, causing MPT. (7) MPT causes stimulation of AIF, which is cleaved by calpain. (8) AIF released in cytoplasm activates programmed cell death (apoptosis). Grp78—immunoglobulin heavy-chain binding protein of heat shock protein 70 family, a regulator of unfolded protein response, which is induced in cells with endoplasmic reticulum stress. They act as a molecular chaperone and a key regulator of Ca2+ homeostasis; GM-CSF improves N-glycosylation and is therefore used as a treatment for congenital neutropenia in JAGN1 mutation. MMP, mitochondrial membrane potential; AIF, apoptosis-inducing factor; JAGN1, jagunal homolog 1 membrane protein; MPT, mitochondrial permeability transition; Ca2+, calcium ion; ROS, reactive oxygen species.
Similar to the case of our patient, who had significantly low IgG levels by 5 months of age, at least two other patients with JAGN1 deficiency have been described as having antibody deficiency (10). In a murine model, JAGN1-deficient B cells had defective antibody production and secretion, altered immunoglobulin glycosylation, and defects in the differentiation and maintenance of plasma cells (11). These findings have been attributed to increased ER stress and dysregulation of the unfolded protein response. Notably, even JAGN1-deficient patients with normal serum immunoglobulin levels demonstrate an altered immunoglobulin glycoprofile when compared with healthy donors, suggesting the need for a very low threshold for immunoglobulin replacement in all patients with JAGN1 deficiency, regardless of the IgG levels (11).
SCN caused by JAGN1 deficiency may be associated with recurrent intracranial hemorrhage, pancreatic insufficiency, failure to thrive, developmental delay, skeletal abnormalities, and recurrent infections (including fungal infections) in the context of erratic neutrophil counts despite treatment with GCSF. In such cases, hematopoietic stem cell transplantation may be considered and is curative (4).
A summary of published case reports with SCN caused by JAGN1 deficiency is presented in Table 3. The case of an index family in Northern Africa was reported, but few details were provided (5). Of the other reported cases, the majority were females either from Turkey or Algeria. The mean age was 10 years and the mean ANC at the time of presentation was 100 cells/µl. Common clinical features associated with the mutation were sepsis, abscess, pneumonia, failure to thrive, and neurodevelopmental delay, and genetic defects were homozygous mutations in exons 1 and 2 of JAGN1 in addition to the alteration of proteins.
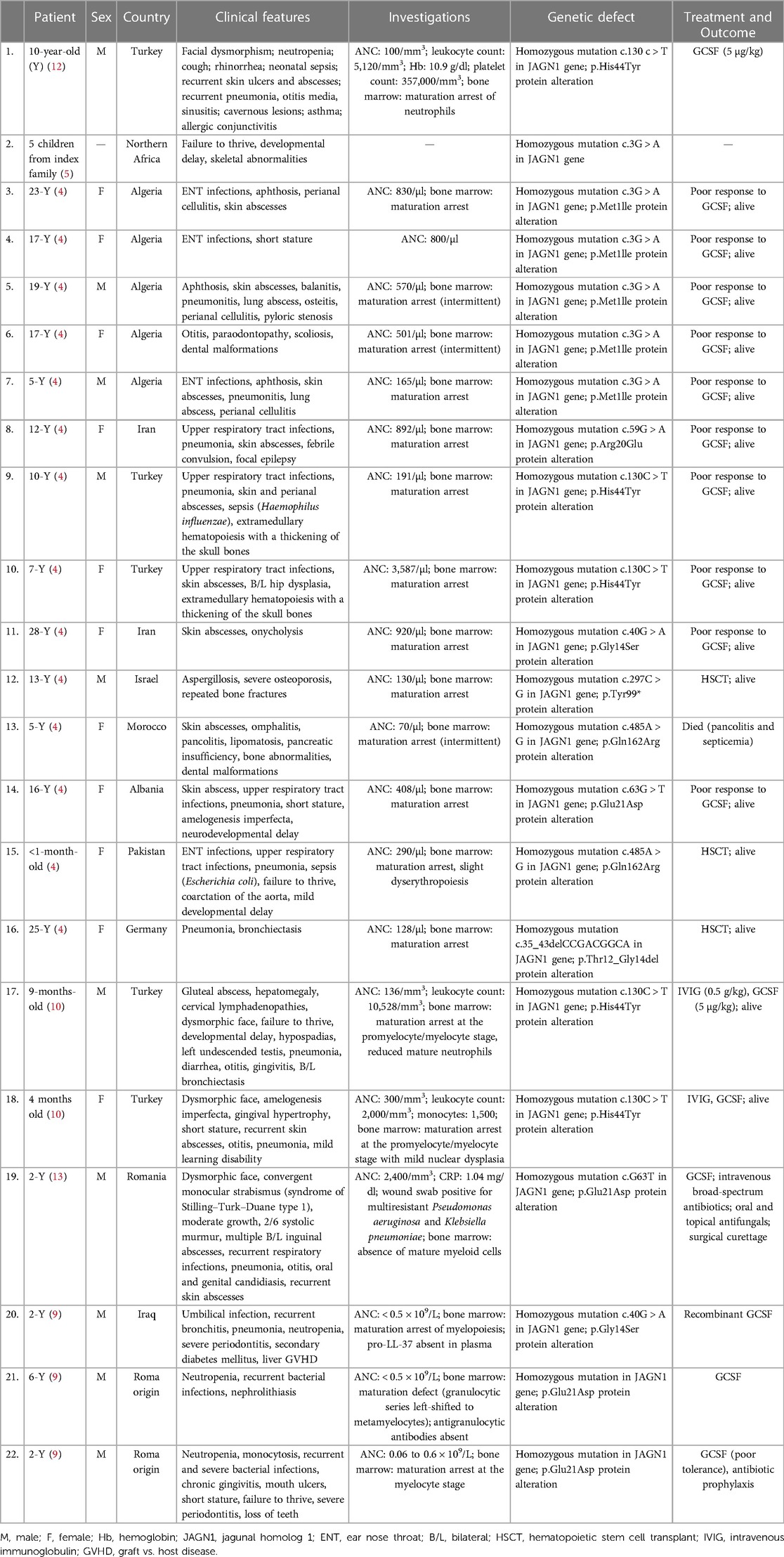
Table 3. Case reports of severe congenital neutropenia caused by the JAGN1 mutation.
Our patient shared some common clinical features with cases reported in the literature, such as recurrent infections during hospital stay. To the best of our knowledge, this is the first report of recurrent intracranial hemorrhage occurring after minimal trauma among patients with JAGN1 deficiency. Our patient was subjected to a thorough evaluation for discovering possible etiologies for his bleeding diathesis. This involved an extensive evaluation for any underlying coagulopathy that was negative (Table 1) as well as platelet function assays. In addition, imaging studies did not reveal any arteriovenous malformations. Despite this thorough outpatient investigation, the etiology of bleeding diathesis remains unknown. It is possible that JAGN1, which enhances glycosylation of neutrophil proteins, has as-yet unknown effects on platelet or coagulation factor function as well.
Conclusion
In this study Here, we report that an invasive epidural abscess caused by P. aeruginosa occurred in a patient with SCN secondary to JAGN1 mutation, who initially presented with periorbital cellulitis. Given the variability seen in the clinical presentation of patients with SCN secondary to JAGN1 deficiency, a high index of suspicion for invasive pyogenic infection should be maintained in those with this immunodeficiency. This should lead to prompt initiation of treatment and prophylaxis for life-threatening infections, especially when patients with SCN secondary to JAGN1 mutation present with new focal neurologic defects, altered mental status, or have new abnormalities noted during physical examination. We also report a history of recurrent intracranial hemorrhage in our patient, which is considered unusual. Bleeding diatheses can be considered a contributor to any new symptom or finding in patients with this condition, and clinicians should consider providing anticipatory guidance and precautions to such patients, which will help achieve the goal of trauma prevention.
Data availability statement
All relevant clinical data are included in the article. Further inquires can be directed to the corresponding author.
Ethics statement
Ethical review and approval were not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent was provided by the patient's legal guardian/next of kin for the publication of this case report.
Author contributions
ST: writing, first and corresponding author, review/proofreading. GG: clinical input, review/proofreading. JR: clinical input, review/proofreading. CP: clinical input, review/proofreading. AS: clinical input, review/proofreading. OL: clinical input, review/proofreading. LG: clinical input, review/proofreading, senior author. All authors contributed to the article and approved the submitted version.
Acknowledgments
We would like to thank the child's family for providing their consent to publish this case report. We are also grateful to Sauradeep Sarkar, MCh., at the Department of Neurosurgery, Brigham and Women's Hospital, for his help with the diagnostic imaging presented in this case report.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Levy O. Antimicrobial proteins and peptides of blood: templates for novel antimicrobial agents. Blood. (2000) 96(8):2664–72. doi: 10.1182/blood.V96.8.2664
2. Gong R-L, Wu J, Chen T-X. Clinical, laboratory, and molecular characteristics and remission status in children with severe congenital and non-congenital neutropenia. Front Pediatr. (2018) 6:305. doi: 10.3389/fped.2018.00305
3. Skokowa J, Dale DC, Touw IP, Zeidler C, Welte K. Severe congenital neutropenias. Nat Rev Dis Primers. (2017) 3:17032. doi: 10.1038/nrdp.2017.32
4. Boztug K, Järvinen PM, Salzer E, Racek T, Mönch S, Garncarz W, et al. JAGN1 deficiency causes aberrant myeloid cell homeostasis and congenital neutropenia. Nat Genet. (2014) 46:1021–7. doi: 10.1038/ng.3069
5. Boztug K, Järvinen PM, Salzer E, Racek T, Mönch S, Garncarz W, et al. Deficiency of JAGN1 causes severe congenital neutropenia associated with defective secretory pathway and aberrant myeloid cell homeostasis. Blood. (2013) 122:439–439. doi: 10.1182/blood.V122.21.439.439
6. Donadieu J, Beaupain B, Fenneteau O, Bellanné-Chantelot C. Congenital neutropenia in the era of genomics: classification, diagnosis, and natural history. Brit J Haematol. (2017) 179:557–74. doi: 10.1111/bjh.14887
7. Wirnsberger G, Zwolanek F, Stadlmann J, Tortola L, Liu SW, Perlot T, et al. Jagunal homolog 1 is a critical regulator of neutrophil function in fungal host defense. Nat Genet. (2014) 46:1028–33. doi: 10.1038/ng.3070
8. Khandagale A, Lazzaretto B, Carlsson G, Sundin M, Shafeeq S, Römling U, et al. JAGN1 Is required for fungal killing in neutrophil extracellular traps: implications for severe congenital neutropenia. J Leukocyte Biol. (2018) 104:1199–213. doi: 10.1002/JLB.4A0118-030RR
9. Khandagale A, Holmlund T, Entesarian M, Nilsson D, Kalwak K, Klaudel-Dreszler M, et al. Severe congenital neutropenia-associated JAGN1 mutations unleash a calpain-dependent cell death programme in myeloid cells. Brit J Haematol. (2021) 192:200–11. doi: 10.1111/bjh.17137
10. Baris S, Karakoc–Aydiner E, Ozen A, Delil K, Kiykim A, Ogulur I, et al. JAGN1 deficient severe congenital neutropenia: two cases from the same family. J Clin Immunol. (2015) 35:339–43. doi: 10.1007/s10875-015-0156-2
11. Hagelkruys A, Wirnsberger G, Stadlmann J, Wöhner M, Horrer M, Vilagos B, et al. A crucial role for Jagunal homolog 1 in humoral immunity and antibody glycosylation in mice and humans. J Exp Med. (2020) 218:e20200559. doi: 10.1084/jem.20200559
12. Çipe FE, Aydoğmuş Ç, Baskın K, Keskindemirci G, Garncarz W, Boztuğ K. A rare case of syndromic severe congenital neutropenia: jagn1 mutation. Turk J Pediatr. (2020) 62:326. doi: 10.24953/turkjped.2020.02.022
Keywords: JAGN1, congenital, neutropenia, children, severe congenital neutropenia (SCN)
Citation: Thomas S, Guenther G, Rowe JH, Platt CD, Shimamura A, Levy O and Ganapathi L (2023) Severe congenital neutropenia due to jagunal homolog 1 (JAGN1) mutation: a case report and literature review. Front. Pediatr. 11:1223191. doi: 10.3389/fped.2023.1223191
Received: 15 May 2023; Accepted: 28 June 2023;
Published: 17 July 2023.
Edited by:
Pilar Giraldo, University of Zaragoza, SpainReviewed by:
Piero Farruggia, ARNAS Ospedali Civico Di Cristina Benfratelli, ItalyAmmar Husami, Cincinnati Children’s Hospital Medical Center, United States
© 2023 Thomas, Guenther, Rowe, Platt, Shimamura, Levy and Ganapathi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sanya Thomas c2FueWEudGhvbWFzQGNoaWxkcmVucy5oYXJ2YXJkLmVkdQ==