 Yu Xiang
Yu Xiang Ying Zhang
Ying Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr., 28 July 2023
Sec. Pediatric Neurology
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1214837
Introduction: Hemiplegic migraine (HM) is a rare subtype of migraine. HM in children may be atypical in the initial stage of the disease, which could easily lead to misdiagnosis.
Methods: We report two cases of atypical hemiplegic migraine that onset as an acute encephalopathy. And a comprehensive search was performed using PubMed, Web of Science, and Scopus. We selected only papers that reported complete clinical information about the patients with CACNA1A or ATP1A2 gene mutation.
Results: Patient #1 showed a de novo mutation, c.674C>A (p. Pro225His), in exon 5 of the CACNA1A gene. And patient #2 showed a missense mutation (c.2143G>A, p. Gly715Arg) in exon 16 of the ATP1A2. Together with our two cases, a total of 160 patients (73 CACNA1A and 87 ATP1A2) were collected and summarized finally.
Discussion: Acute encephalopathy is the main manifestation of severe attacks of HM in children, which adds to the difficulty of diagnosis. Physicians should consider HM in the differential diagnosis of patients presenting with somnolence, coma, or convulsion without structural, epileptic, infectious, or inflammatory explanation. When similar clinical cases appear, gene detection is particularly important, which is conducive to early diagnosis and treatment. Early recognition and treatment of the disease can help improve the prognosis.
Hemiplegic migraine (HM) is a rare subtype of migraine characterized by episodes of severe migraine and aura symptoms involving motor weakness or numbness, usually affecting one side of the body (hemiparesis), as well as visual, sensory, or speech disturbances (1). Severe attacks can be accompanied by seizures, coma, encephalopathy, fever, cerebellar involvement, cerebral edema, or cerebral infarction (2) and are often misdiagnosed as having postictal confusion, Todd’s paresis, and diagnosed as having epilepsy or viral encephalitis.
We report two cases of atypical hemiplegic migraine that started as an acute encephalopathy. The atypical clinical presentation of these two patients led to a challenging diagnosis. Therefore, we are reporting it to deepen the understanding of this disease. The goal is to reduce misdiagnosis, give patients appropriate and effective treatment, and improve the quality of their survival.
Blood samples (2 ml) were collected from patients and their parents. DNA was isolated from peripheral blood using a DNA Isolation Kit (Blood DNA Kit V2, CW2553). Concentrations were determined on a Qubit fluorometer (Invitrogen, Q33216) using a Qubit dsDNA HS Assay Kit (Invitrogen, Q32851). Agarose gel (1%) electrophoresis was performed for quality control. DNA libraries were prepared with a KAPA Library Preparation Kit (Kapa Biosystems, KR0453) following the manufacturer's instructions. Hybridization of pooled libraries to the capture probes (IDT xGen Exome Research Panel v1.0) and removal of non-hybridized library molecules were carried out according to the SeqCap Hybrid Mix System. Sample dilution, flow cell loading, and sequencing were performed according to the Illumina specifications. DNA libraries were sequenced on the Illumina NovaSeq platform as paired-end 200-bp reads.
Sequence variants were annotated using population and literature databases, including 1000 Genomes (http://www.1000genomes.org/), dbSNP (https://www.ncbi.nlm.nih.gov/snp/), GnomAD (http://gnomad.broadinstitute.org/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), HGMD (http://www.hgmd.cf.ac.uk/ac/index.php), and Online Mendelian Inheritance in Man (OMIM) (http://omim.org/). The variant interpretation was performed according to the American College of Medical Genetics (ACMG) guidelines.
Patient #1, a 3-year-old boy, was the second child of his unrelated parents. He was born via a normal pregnancy and delivery. His mother has a history of migraine. He was admitted to the Affiliated Hospital of Qingdao University because of headache, somnolence, status epilepticus, and fever for half a day.
The patient experienced a total of three similar episodes since the age of 1 year and 7 months. The patient's mental and motor development was normal before and after the onset of the disease. Each time, there was a minor head trauma, followed by headache, somnolence, and seizures. Each seizure manifested as a double-eye gaze to the left, intermittent left-sided limb rigidity, and unconsciousness. Each seizure manifested as status epilepticus. Half a day after the resolution of this seizure, the patient developed a fever with a temperature of 39°C. There were no abnormalities in brain magnetic resonance imaging (MRI), magnetic resonance angiography (MRA), and magnetic resonance venography (MRV) during previous episodes. The blood routine, arterial blood gas analysis, blood ammonia, glucose, homocysteine, calcium, and electrolytes showed no significant abnormalities. A brain MRI after this episode showed multiple high signal intensities on diffusion-weighted imaging (DWI) in the right frontotemporal cortex (Figure 1). Neurological examination after this episode showed the patient had left-sided central facial palsy as well as left-sided upper and lower extremity paralysis. Babinski's sign was positive bilaterally, the rest of the pathological symptoms were negative, and the meningeal stimulation sign was negative.
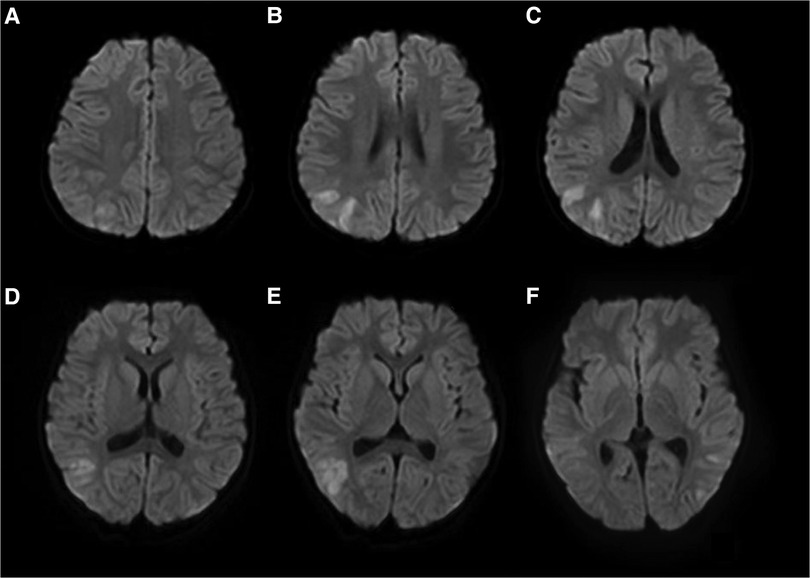
Figure 1. Brain MRI of patient #1 showed multiple high signal intensities on DWI in the right frontotemporal cortex (A–F).
The cerebrospinal fluid (CSF) examination revealed that the pressure, glucose, chloride, protein level, cell count, and classification were all normal. The bacteria and fungi of CSF were negative. The peripheral blood and CSF demyelinating associated antibodies (MOG, AQP4, MBP) were all negative. Video electroencephalogram (EEG) monitoring showed persistent asymmetry between the right and left hemispheres and slow and sharp slow-wave emission in the right hemisphere. The absence of physiological waves in the right hemisphere is shown in Figure 2. During the same period, a focal seizure was monitored which lasted about 90 s (Figure 3).
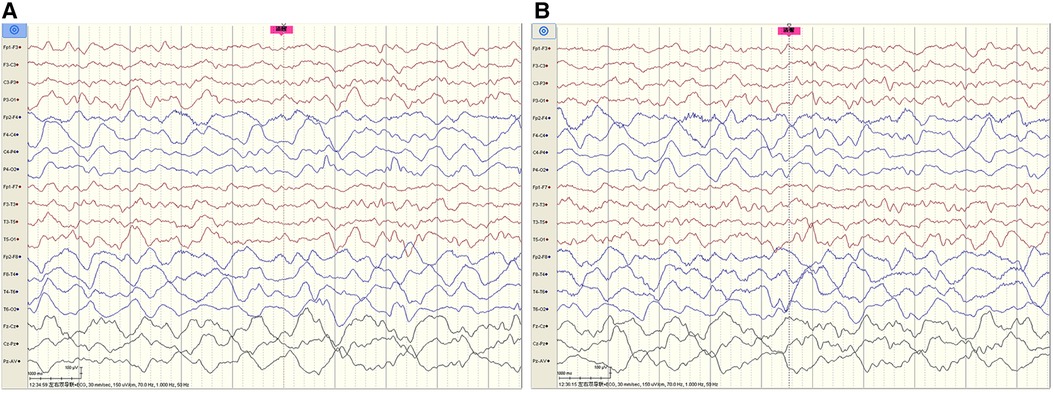
Figure 2. Video EEG monitoring of patient #1 showed persistent asymmetry between the right and left hemispheres and slow and sharp-slow wave emission in the right hemisphere. The absence of physiological waves in the right hemisphere is shown in A, B. The fuchsia rectangle above A, B indicates that Patient #1 is beginning to wake up.
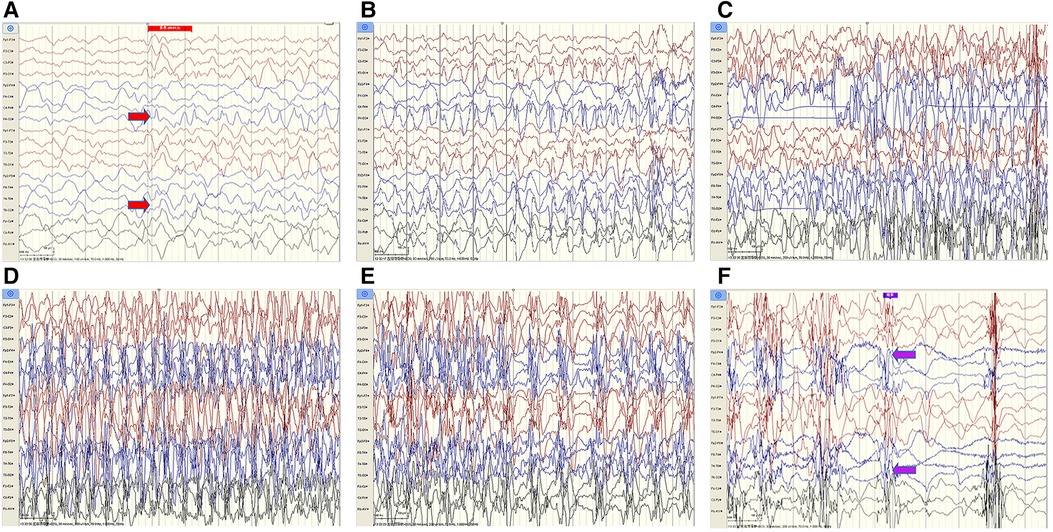
Figure 3. Patient #1 captured a focal seizure during video-EEG monitoring (A–F). Seizures started at the red arrow and stopped at the purple arrow for a total duration of about 90 seconds.
Combining the clinical manifestations of the patient's three attacks, we considered that the patient had a high probability of HM presented as an acute encephalopathy. In addition to conventional fluid and nutritional support therapy, the patient was given methylprednisolone (3 mg/kg/d) to reduce cerebral edema and mannitol (2.6 g/kg/d) to lower cranial pressure. Flunarizine (0.2 mg/kg/d) was given to dilate cerebral blood vessels. Disturbances in the patient's consciousness were ameliorated gradually. Left hemiplegia also gradually resolved, and he recovered completely without sequelae 9 days after the onset. The patient's review of video EEG monitoring on day 11 showed asymmetric background activity in the right and left occipital regions, with slow-wave coverage on the right side and predominantly slow and sharp slow-wave emission in the right hemisphere (Figure 4).
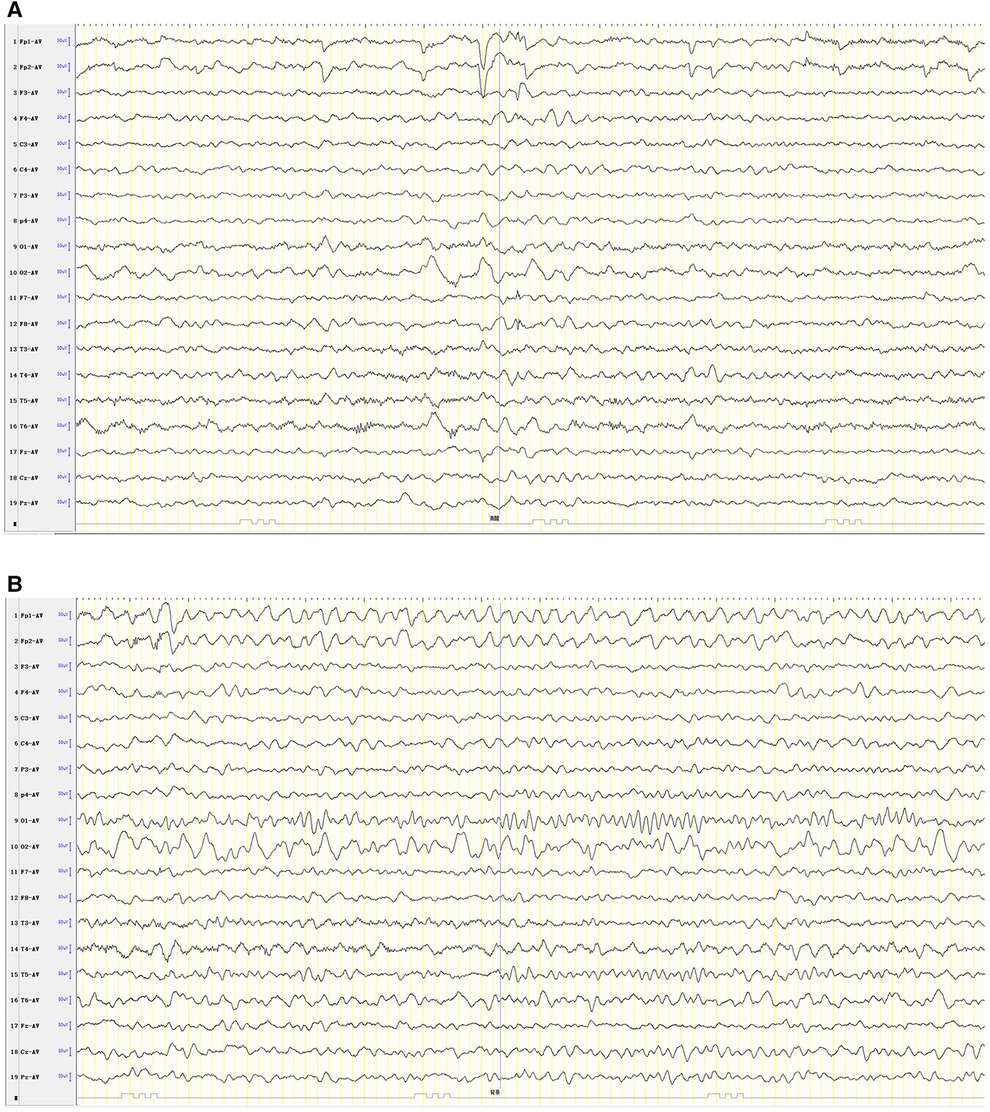
Figure 4. Patient #1 was reexamined for video EEG monitoring on day 11 of illness (A, B).
After communicating with the patient's family, whole-exome sequencing (WES) was performed in the Running Gene Medical laboratory in Beijing, China. WES found a de novo mutation, c.674C>A (p. Pro225His), in exon 5 of the CACNA1A gene (Figure 5). Neither of his parents had the mutation. This mutation was predicted to be disease-causing by Mutation Taster (http://www.mutationtaster.org/) and was predicted to be damaging with a score of 1.000 (sensitivity: 0.00; specificity: 1.00) by Polyphen 2 (http://genetics.bwh.harvard.edu/pph2/) and damaging with a score of 0.000 by SIFT (cutoff = 0.05) (http://sift.jcvi.org/www/SIFT_BLink_submit.html).

Figure 5. Patient #1 and his parents’ whole-exome sequencing results. Patient #1 has a de nove mutation of c.674C>A (p. Pro225His), in exon 5 of the CACNA1A gene. Neither of his parents carried the disease-modifying mutation. The circles indicate the locus of variation.
The patient was finally diagnosed with hemiplegic migraine presenting as an acute encephalopathy. He was given daily flunarizine 3 mg (0.2 mg/kg) to prevent HM attacks. After treatment for 1.5 months, a follow-up MRI showed complete disappearance of abnormal signals throughout the right hemispheric cortex (Figure 6). The video EEG was not reviewed because the patient fully recovered to the pre-onset level and the patient's compliance was not high.

Figure 6. The brain MRI of Patient #1 showed complete disappearance of abnormal signals 1.5 months after treatment (A–D).
Patient #2 was a 5-year-old boy who was the second child of his unrelated parents. He was admitted to the Affiliated Hospital of Qingdao University because of focal seizures and somnolence for half a day. The seizure was characterized by double-eye gaze, cyanosis of the lips, and shaking upper limbs, which lasted for about 1 min. The body temperature was 38°C. The blood routine, blood electrolytes, ammonia, glucose, and calcium were all normal. Brain CT showed no significant abnormalities. Neurological examination showed a Glasgow Coma Scale of 8. The patient's extremity muscle strength and muscle tone were normal. Babinski's sign was positive bilaterally, while the rest of the pathological signs were negative, and the meningeal stimulation sign was negative.
The patient was born via a normal pregnancy and delivery. He presented with previously regular intellectual and motor development. He started to have febrile seizures at the age of 1 year and had a total of three times by the age of 2 years. He had a history of viral encephalitis 3 years ago. He suffered a headache, nausea, and vomiting after mild head trauma 2 years ago. The result of the brain CT was normal. Finally, the symptoms were relieved after rest. His mother had a history of seizures. His sister was diagnosed with epilepsy and was being treated with valproic acid. She also had a history of seizures and coma after mild head trauma.
The examination was immediately completed after admission. Blood biochemistry, blood homocysteine, blood culture, and herpes simplex virus test were all normal. A brain MRI was not performed on admission because the patient was uncooperative. Considering the patient's family history, we recommended that the patient complete the WES, but the family declined.
The patient was given a combination of mannitol (2 g/kg/d), methylprednisolone (2 mg/kg/d), meropenem (60 mg/kg/d), acyclovir (27 mg/kg/d), and vitamin B6 (6 mg/kg/d). On the second day of admission, the patient had another seizure, which lasted about 4 min. The body temperature was 38.2°C. He was treated with midazolam and completed a lumbar puncture. The biochemical and cytologic examination of the CSF revealed normal glucose, chloride, protein level, cell count, and classification. The CSF tests for bacteria, viruses, and fungi were all negative. The patient was continued to be given an ice blanket and ice cap to lower the temperature and combination therapy. On the fourth day of admission, the patient's temperature gradually decreased to normal, and no further seizures occurred. His consciousness gradually improved with continued treatment as described above. A brain MRI was performed on the 13th day of admission, and the results were normal. On the 14th day of admission, the patient recovered to the pre-onset level, submitted whole-exome sequencing, and was discharged.
WES was performed by Fujun Genetics Medical Laboratory, Fuzhou, China. The result showed a missense mutation c.2143G>A (p. Gly715Arg) in exon 16 of the ATP1A2 gene (Figure 7) previously reported as a pathogenic mutation (3, 4). The same mutations were confirmed in the patient's mother and sister (Figure 7). This mutation was predicted to be disease-causing by Mutation Taster (http://www.mutationtaster.org/) and was predicted to be damaging with a score of 1.000 (sensitivity: 0.00; specificity: 1.00) by Polyphen 2 (http://genetics.bwh.harvard.edu/pph2/) and damaging with a score of 0.000 by SIFT (cutoff = 0.05) (http://sift.jcvi.org/www/SIFT_BLink_submit.html).
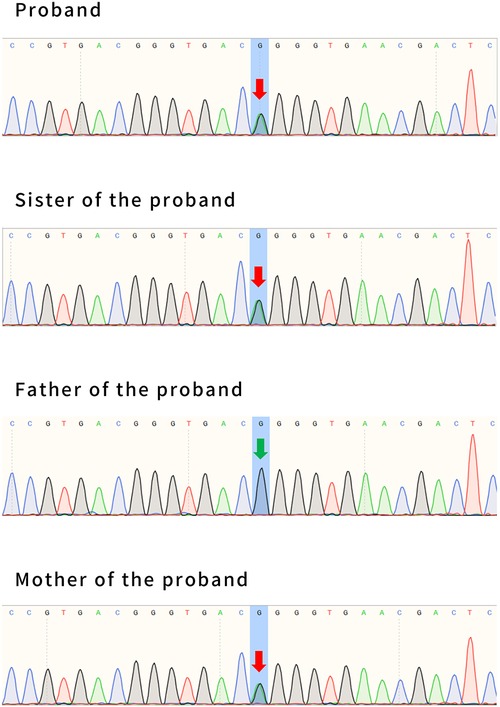
Figure 7. The whole-exome sequencing results of patient #2, his sister, and his parents. Patient #2 has a maternal mutation of c.2143G>A (p. Gly715Arg), in exon 16 of the ATP1A2 gene. The same mutations were confirmed in his sister. His father had no mutation at this locus. The arrows indicate the locus of variation.
Based on a series of examinations, the patient was finally diagnosed with acute encephalopathy of HM. He was subsequently treated with flunarizine at 5 mg/day.
The overall estimated prevalence of HM is 0.01% (5). The prevalence of sporadic HM (SHM) is at least 0.002% and the prevalence of familial HM (FHM) is at least 0.003% based on the Danish population (6–8). The onset is usually in the first or second decade (9). The frequency, intensity, and duration of HM attacks often decrease with age (5, 8). Emotional and intense physical stress, viral infections, and head trauma are the more commonly reported trigger factors for HM attacks (10). The two patients in this paper were both male and the age of onset was all before 5 years of age. For patient #1, the trigger was mild head trauma. The trigger for Patient #2 is unclear, and the child had runny nose before this episode, so we hypothesized that it might be related to a viral infection.
HM can be divided into FHM and SHM (11). If one first- or second-degree relative has similar symptoms, the disorder can be classified as FHM. Otherwise, it is classified as SHM (11). FHM can be classified as FHM1 (OMIM #141500), FHM2 (OMIM #602481), or FHM3 (OMIM #609634) according to mutations in CACNA1A, ATP1A2, or SCN1A, respectively (1, 5, 11). SHM can be caused by a de novo mutation in a gene that causes the familial form or by the inheritance of a gene mutation from an asymptomatic parent with FHM (12). A review study by Bonemazzi et al. found that pediatric SHM patients have longer and more severe attacks compared to FHM patients, especially during the initial years after disease onset, while FHM cases tend to have the disease for longer periods (8).
A comprehensive search was performed using PubMed, Web of Science, and Scopus. We selected only papers that reported complete clinical information about the patients with CACNA1A or ATP1A2 gene mutation. We screened 97 articles finally. Together with our two cases, a total of 160 patients (73 CACNA1A and 87 ATP1A2) were identified and all features are summarized in Table 1.
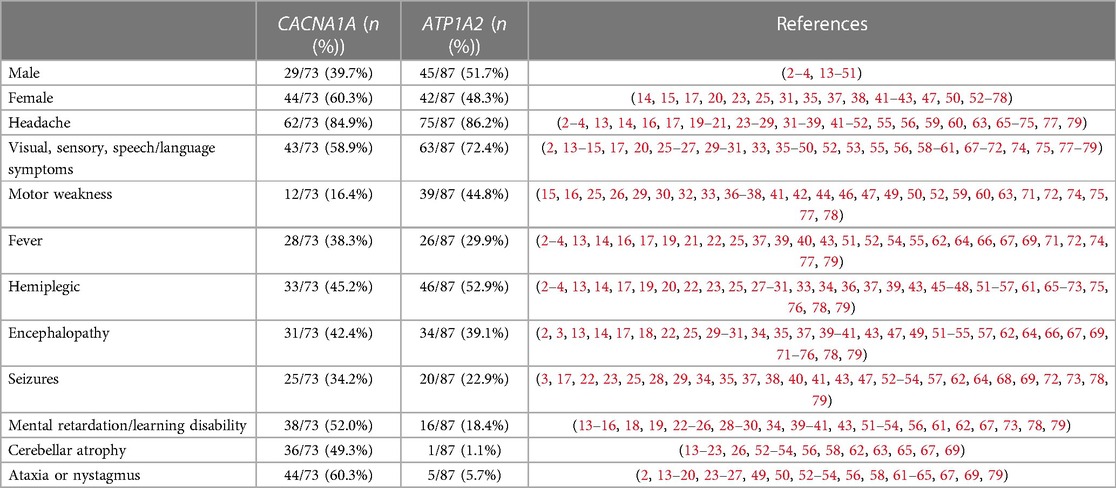
Table 1. Clinical characteristics of HM patients with CACNA1A or ATP1A2 gene mutation.
We found that about half of them presented with a typical hemiplegic migraine attack, i.e., fully reversible visual, sensory, or speech symptoms or motor weakness. Motor symptoms are most frequently localized in hands, arms, feet, legs, tongue, face, and body (8). The progression of weakness is always gradual, requiring at least 5 min, and the irradiation of the symptom is usually unilateral configuring a hemiparesis in most cases (8). Duration is about 30 min to 24 h (mean duration 5 h) for motor aura (8). Of the two patients we reported, patient #1 presented with hemiparesis symptoms that lasted more than 24 h, which is inconsistent with the description of motor aura symptoms in previous reports. Both patients reported in this paper did not describe any other aura symptoms, such as sensory aura, visual aura, or aphasic aura, which may be related to the patient's young age and lack of expressive skills.
However, some patients started with other atypical symptoms, such as encephalopathy. Of the reported patients with hemiplegic migraine with CACNA1A mutations, 42.4% presented with encephalopathy. Among patients with ATP1A2 mutations, 39.1% presented with encephalopathy. The specific manifestations of encephalopathy were impaired level of consciousness, fever, or seizures, all lasting for >24 h. Patients with mild encephalopathy have only an impaired level of consciousness, while patients with severe manifestations of encephalopathy may have fever and seizures. In this paper, we found that all patients presenting with fever showed signs of encephalopathy. Fever may be central (79). In our group, both probands had a sudden onset of fever without a clear history of infection, and neither antibiotic nor antipyretic was effective for them. The clinical manifestations began to improve after the body temperature returned to normal in both probands. Both our patients manifested focal seizures followed by generalized seizures, of which patient #1 started with status epilepticus in each case. The whole-exome sequencing of patient #1 showed a de novo missense mutation of the CACNA1A gene, c.674C>A (p. Pro825His). By reviewing the literature, only one report of the Pro225His mutation was identified (80). The patient was an 8-year-old female who was born prematurely, and presented with developmental delay, speech and spatial sense impairment, poor concentration, and epilepsy (80). The patient had at least four HM episodes presenting as headaches and seizures (80). The difference is that our patient #1 was born at full term and had normal cognitive and motor development. He had no previous seizures. Similarly, our patient presented this time with status epilepticus and hemiplegia. However, we are currently unable to clarify the clinical phenotype of these mutations due to the paucity of reports. The whole-exome sequencing of patient #2 showed a maternally inherited missense mutation of the ATP1A2 gene, c.2143G>A (p. Gly715Arg). By reviewing the literature, three patients have been reported thus far with the same mutations as patient #2. Together with our case, Table 2 summarizes the clinical features of these four patients. All four patients had their onset in childhood and their age of onset ranged from 1 to 6 years, with our patient #2 having the youngest age of onset. However, because all four patients started with acute encephalopathy, this led to different degrees of diagnostic delay. Patients may present with fever, somnolence, hemiparesis, seizures, or aphasia during the acute phase of encephalopathy. All three patients showed cerebral edema in the acute phase, except for our patient #2 for whom no brain MRI data were in the acute phase. Patients’ encephalopathy symptoms may resolve within a few weeks, with recovery in up to 10 weeks. Combined with the clinical presentation of these four patients, we hypothesized that this mutation may be associated with early and severe HM manifestations.

Table 2. Summary of HM caused by ATP1A2, c.2143G>A mutation.
The mechanism of HM accompanied by acute encephalopathy is still unknown. Toldo et al. reported an 8-year-old female who had a prolonged attack of sporadic HM; the first MRI was negative while the neuroradiological follow-up documented a progressive increase of the cortical swelling with mild hyperintensity on DWI, suggesting intracellular edema (81). Technetium-99m-ethyl cysteinate dimmer (99mTc-ECD) single photon emission computed tomography (SPET) (day 27), performed when the hemiplegia was almost completely resolved but aphasia persisted, showed a marked unilateral cerebral hypoperfusion. This finding suggested a primary neuronal dysfunction with reduced uptake of the radiopharmaceutical by “stunned neuronal cells” (81). The neuroimaging findings have been related to the possible underlying pathogenic mechanisms, ion channel dysfunction caused prolonged neuronal depolarization followed by the shift of water from the extra- to the intra-cell compartment and then cellular swelling and neuronal loss (81).
The outcome is favorable in most patients; however, neurological conditions and comorbidities can be associated (8). Some studies reported neurological signs in up to 60% of patients, with the most frequent neurological alterations including cerebellar ataxia, nystagmus, postural tremor, and clumsiness (8). The majority of FHM1 patients, often more than half of the families (2), and a small number of FHM2 patients had cerebellar signs, and cerebellar atrophy was seen on brain MRI (82). Cerebellar atrophy may become apparent with age (83). In our group, both patients experienced multiple hemiplegic migraine attacks, but neither of them showed cerebellar signs, cerebellar atrophy, motor development lag, or regression, but it cannot be excluded that this is related to the young age of both patients and long-term follow-up is needed. In addition, Wada et al. considered that the mutation of CACNA1A itself, rather than the frequency of coma attacks, may play a central role in the pathogenesis of progressive cerebellar atrophy (14). Cerebellar signs are rare in patients with FHM2. Three patients reported by Spadaro et al. showed cerebellar symptoms such as nystagmus, gait ataxia, dysmetria, and dysarthria, one of the patients also showed cerebellar atrophy (69). It was thought that the presentation may be related to the patient's alcohol or sedative intake (84). However, in other families, no other reasons for the cerebellar signs such as alcohol, drugs, CACNA1A or SCA mutations, or vascular brain disorders could be found, suggesting that these signs are part of the FHM2 phenotype in this family (69).
We found that about half of the HM patients with the CACAN1A gene mutation had mental retardation, which may be related to the calcium channel impairment caused by the CACNA1A gene mutation. For our patient #1 with CACNA1A gene mutation, he experienced three hemiplegic migraine attacks, and no mental retardation or regression manifestation has been found yet, which may be related to the patient's young age or insufficient follow-up time. Observation of this patient should continue to be strengthened in the future. Overall, more research is needed to explore the underlying mechanisms of acute encephalopathy in HM.
At present, the impact of the ATP1A2 mutation on cognitive profile in HM patients has not been evaluated in detail (40). In this paper, we found that most of the effects of the ATP1A2 gene on intelligence were transient and could gradually return to pre-initiation levels after the remission of HM symptoms. However, Wang et al. reported a patient with cognitive dysfunction in a specific area (40). He had trouble in mathematics and depicting three-dimensional things. It is suggested that a patient with HM with ATP1A2 could develop permanent cognitive dysfunction (40). As for our patient #2, a careful review of his history revealed that the patient and his sister were normal in all aspects except for poor numeracy. The patient's mother had cognitive impairment, but she refused to undergo intelligence testing. The cognitive function test is an issue we need to focus on when following up with patient #2 in the future.
Patients with HM need only symptomatic treatment in the acute phase to bring about symptom relief. Some reports indicated that a prompt recovery had been described in three patients with CACNA1A mutations through the combined use of steroids and hypertonic solutions in the course of encephalopathy and cerebral edema (54, 83, 85). For FHM2, NMDA receptor antagonist memantine could prevent glutamatergic excitotoxicity, and idebenone, dl-NBP, as well as traditional anti-migraine drugs could protect the mitochondria function (73). We report two patients who started with acute encephalopathy and were treated symptomatically in the acute phase. Both patients were given low-dose methylprednisolone combined with mannitol. No drugs such as memantine and idebenone were applied to patient #2 because the diagnosis had not been established before. The WES results can help us select more effective medication in the future.
Prophylactic management is applied to patients with frequent, long-lasting, or severe attacks (12). A single-center cohort study of pediatric migraine lasting 11 years suggested that flunarizine should be used as first-line medication in children with hemiplegic migraine (86). In the two patients we reported, considering the patients’ age, weight, and tolerance to the drug, the dose of flunarizine was 0.2 mg/kg per day, with a maximum amount of 5 mg/d. The two patients we reported were treated with flunarizine for 8and 2 months, respectively, and both were well tolerated without adverse effects. During the current follow-up, patient #1 had occasional headaches that could be resolved with rest. He has not suffered any head trauma and has not undergone severe attacks such as convulsions or encephalopathy. He is still under continued follow-up. Patient #2 did not experience any further headaches or convulsions, probably due to the short follow-up period, and is still under continued follow-up.
Acute encephalopathy is the main manifestation of severe attacks of HM in children, which adds to the difficulty of diagnosis. Physicians should consider HM in the differential diagnosis of patients presenting with somnolence, coma, or convulsion without structural, epileptic, infectious, or inflammatory explanation. Early recognition and treatment of the disease can help improve the prognosis. Whole-exome sequencing can suggest diagnosis and the type. Future multicenter and large sample size studies are still needed to explore the treatment strategy for this disease.
Original datasets are available in the NCBI repository: The original contributions presented in the study are publicly available. The BioProject ID is PRJNA993808. This data can be found here: http://www.ncbi.nlm.nih.gov/bioproject/993808.
The studies involving human participants were reviewed and approved by the Affiliated Hospital of Qingdao University (approval number: QYFYWZLL27854) and the 1964 Declaration of Helsinki. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
All authors contributed to the study's conception and design. The investigation, validation, and writing—original draft were performed by YX. Data curation and formal analysis were performed by ZS, ZY, and CY. Writing—review and editing was performed by FL. Supervision, resources, and project administration were performed by JX and YZ. All authors contributed to the article and approved the submitted version.
This work was supported by the Taishan Scholars Program of Shandong Province (no. tsqn201909191), the Youth Fund of the Affiliated Hospital of Qingdao University (no. QDFYQN2020014), and the Youth Fund of Shandong Natural Science Foundation (no. ZR2021QH042) in the collection, analysis, and interpretation of data and in writing the manuscript.
We thank the patients and their parents for their kind cooperation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sutherland HG, Albury CL, Griffiths LR. Advances in genetics of migraine. J Headache Pain. (2019) 20(1):72. doi: 10.1186/s10194-019-1017-9
2. Romozzi M, Primiano G, Rollo E, Travaglini L, Calabresi P, Servidei S, et al. CACNA1A-p.Thr501Met mutation associated with familial hemiplegic migraine: a family report. J Headache Pain. (2021) 22(1):85. doi: 10.1186/s10194-021-01297-5
3. Chen H, Sun X, Wang R, Yi Z, Huang Z, Xie J, et al. A case report of atypical hemiplegic migraine with nonheadache onset in a Chinese child. BMC Neurol. (2021) 21(1):267. doi: 10.1186/s12883-021-02302-9
4. De Sanctis S, Grieco GS, Breda L, Casali C, Nozzi M, Del Torto M, et al. Prolonged sporadic hemiplegic migraine associated with a novel de novo missense ATP1A2 gene mutation. Headache. (2011) 51(3):447–50. doi: 10.1111/j.1526-4610.2010.01793.x
5. Di Stefano V, Rispoli MG, Pellegrino N, Graziosi A, Rotondo E, Napoli C, et al. Diagnostic and therapeutic aspects of hemiplegic migraine. J Neurol Neurosurg Psychiatry. (2020) 91(7):764–71. doi: 10.1136/jnnp-2020-322850
6. Eriksen MK, Thomsen LL, Olesen J. Implications of clinical subtypes of migraine with aura. Headache. (2006) 46(2):286–97. doi: 10.1111/j.1526-4610.2006.00286.x
7. Thomsen LL, Ostergaard E, Olesen J, Russell MB. Evidence for a separate type of migraine with aura: sporadic hemiplegic migraine. Neurology. (2003) 60(4):595–601. doi: 10.1212/01.wnl.0000046524.25369.7d
8. Bonemazzi I, Brunello F, Pin JN, Pecoraro M, Sartori S, Nosadini M, et al. Hemiplegic migraine in children and adolescents. J Clin Med. (2023) 12(11):3783. doi: 10.3390/jcm12113783
9. Indelicato E, Boesch S. From genotype to phenotype: expanding the clinical Spectrum of CACNA1A variants in the era of next generation sequencing. Front Neurol. (2021) 12:639994. doi: 10.3389/fneur.2021.639994
10. Toldo I, Brunello F, Morao V, Perissinotto E, Valeriani M, Pruna D, et al. First attack and clinical presentation of hemiplegic migraine in pediatric age: a multicenter retrospective study and literature review. Front Neurol. (2019) 10:1079. doi: 10.3389/fneur.2019.01079
11. Headache classification committee of the international headache society (IHS). The international classification of headache disorders, 3rd edition. Cephalalgia. (2018) 38(1):1–211. doi: 10.1177/0333102417738202
12. Russell MB, Ducros A. Sporadic and familial hemiplegic migraine: pathophysiological mechanisms, clinical characteristics, diagnosis, and management. Lancet Neurol. (2011) 10(5):457–70. doi: 10.1016/s1474-4422(11)70048-5
13. Stam AH, Vanmolkot KR, Kremer HP, Gärtner J, Brown J, Leshinsky-Silver E, et al. CACNA1A R1347q: a frequent recurrent mutation in hemiplegic migraine. Clin Genet. (2008) 74(5):481–5. doi: 10.1111/j.1399-0004.2008.00996.x
14. Wada T, Kobayashi N, Takahashi Y, Aoki T, Watanabe T, Saitoh S. Wide clinical variability in a family with a CACNA1A T666m mutation: hemiplegic migraine, coma, and progressive ataxia. Pediatr Neurol. (2002) 26(1):47–50. doi: 10.1016/s0887-8994(01)00371-x
15. Freilinger T, Bohe M, Wegener B, Müller-Myhsok B, Dichgans M, Knoblauch H. Expansion of the phenotypic spectrum of the CACNA1A T666M mutation: a family with familial hemiplegic migraine type 1, cerebellar atrophy and mental retardation. Cephalalgia. (2008) 28(4):403–7. doi: 10.1111/j.1468-2982.2008.01540.x
16. Freilinger T, Ackl N, Ebert A, Schmidt C, Rautenstrauss B, Dichgans M, et al. A novel mutation in CACNA1A associated with hemiplegic migraine, cerebellar dysfunction and late-onset cognitive decline. J Neurol Sci. (2011) 300(1–2):160–3. doi: 10.1016/j.jns.2010.09.032
17. Chan YC, Burgunder JM, Wilder-Smith E, Chew SE, Lam-Mok-Sing KM, Sharma V, et al. Electroencephalographic changes and seizures in familial hemiplegic migraine patients with the CACNA1A gene S218l mutation. J Clin Neurosci. (2008) 15(8):891–4. doi: 10.1016/j.jocn.2007.01.013
18. García-Baró-Huarte M, Iglesias-Mohedano AM, Slöcker-Barrio M, Vázquez-López M, García-Morín M, Miranda-Herrero MC, et al. Phenotypic variability in a four generation family with a p.Thr666Met CACNA1A gene mutation. Pediatr Neurol. (2014) 51(4):557–9. doi: 10.1016/j.pediatrneurol.2014.07.008
19. Frusciante R, Capuano A, Travaglini L, Zanni G, Vigevano F, Bertini E, et al. P016. Congenital ataxia, hemiplegic migraine due to a novel mutation of CACNA1A: a case report. J Headache Pain. (2015) 16(Suppl 1):A146. doi: 10.1186/1129-2377-16-s1-a146
20. Khaiboullina SF, Mendelevich EG, Shigapova LH, Shagimardanova E, Gazizova G, Nikitin A, et al. Cerebellar atrophy and changes in cytokines associated with the CACNA1A R583Q mutation in a Russian familial hemiplegic migraine type 1 family. Front Cell Neurosci. (2017) 11:263. doi: 10.3389/fncel.2017.00263
21. Tashiro Y, Yamazaki T, Nagamine S, Mizuno Y, Yoshiki A, Okamoto K. Repeated encephalopathy and hemicerebral atrophy in a patient with familial hemiplegic migraine type 1. Intern Med. (2014) 53(19):2245–50. doi: 10.2169/internalmedicine.53.0295
22. Ohmura K, Suzuki Y, Saito Y, Wada T, Goto M, Seto S. Sporadic hemiplegic migraine presenting as acute encephalopathy. Brain Dev. (2012) 34(8):691–5. doi: 10.1016/j.braindev.2011.11.002
23. Stam AH, Luijckx GJ, Poll-Thé BT, Ginjaar IB, Frants RR, Haan J, et al. Early seizures and cerebral oedema after trivial head trauma associated with the CACNA1A S218l mutation. J Neurol Neurosurg Psychiatry. (2009) 80(10):1125–9. doi: 10.1136/jnnp.2009.177279
24. Naik S, Pohl K, Malik M, Siddiqui A, Josifova D. Early-onset cerebellar atrophy associated with mutation in the CACNA1A gene. Pediatr Neurol. (2011) 45(5):328–30. doi: 10.1016/j.pediatrneurol.2011.08.002
25. Asghar SJ, Milesi-Hallé A, Kaushik C, Glasier C, Sharp GB. Variable manifestations of familial hemiplegic migraine associated with reversible cerebral edema in children. Pediatr Neurol. (2012) 47(3):201–4. doi: 10.1016/j.pediatrneurol.2012.05.006
26. Gandini MA, Souza IA, Ferron L, Innes AM, Zamponi GW. The de novo CACNA1A pathogenic variant Y1384C associated with hemiplegic migraine, early onset cerebellar atrophy and developmental delay leads to a loss of Cav2.1 channel function. Mol Brain. (2021) 14(1):27. doi: 10.1186/s13041-021-00745-2
27. Kierdaszuk B, Dziewulska D, Pronicka E, Trubicka J, Rokicki D, Piekutowska-Abramczuk D, et al. Identification of the first in Poland CACNA1A gene mutation in familial hemiplegic migraine. Case report. Neurol Neurochir Pol. (2017) 51(2):184–9. doi: 10.1016/j.pjnns.2017.01.005
28. Wilbur C, Buerki SE, Guella I, Toyota EB, Evans DM, McKenzie MB, et al. An infant with epilepsy and recurrent hemiplegia due to compound heterozygous variants in ATP1A2. Pediatr Neurol. (2017) 75:87–90. doi: 10.1016/j.pediatrneurol.2017.06.003
29. Zhang HY, Ma JH, Sun D. Sporadic hemiplegic migraine due to a missense variation in ATP1A2 gene in 2 children. Zhonghua Er Ke Za Zhi. (2022) 60(6):594–5. doi: 10.3760/cma.j.cn112140-20220110-00032
30. Cobb-Pitstick K, Cummings DD, Zuccoli G. Prolonged hyperperfusion in a child with ATP1A2 defect-related hemiplegic migraine. Can J Neurol Sci. (2020) 47(5):687–8. doi: 10.1017/cjn.2020.83
31. Pierelli F, Grieco GS, Pauri F, Pirro C, Fiermonte G, Ambrosini A, et al. A novel ATP1A2 mutation in a family with FHM type II. Cephalalgia. (2006) 26(3):324–8. doi: 10.1111/j.1468-2982.2006.01002.x
32. Montani D, Girerd B, Günther S, Riant F, Tournier-Lasserve E, Magy L, et al. Pulmonary arterial hypertension in familial hemiplegic migraine with ATP1A2 channelopathy. Eur Respir J. (2014) 43(2):641–3. doi: 10.1183/09031936.00147013
33. Pelzer N, Stam AH, Carpay JA, Vries BD, van den Maagdenberg AM, Ferrari MD, et al. Familial hemiplegic migraine treated by sodium valproate and lamotrigine. Cephalalgia. (2014) 34(9):708–11. doi: 10.1177/0333102413520086
34. Podestà B, Briatore E, Boghi A, Marenco D, Calzolari S. Transient nonverbal learning disorder in a child suffering from familial hemiplegic migraine. Cephalalgia. (2011) 31(14):1497–502. doi: 10.1177/0333102411418260
35. Pelzer N, Blom DE, Stam AH, Vijfhuizen LS, Hageman A, van Vliet JA, et al. Recurrent coma and fever in familial hemiplegic migraine type 2. A prospective 15-year follow-up of a large family with a novel ATP1A2 mutation. Cephalalgia. (2017) 37(8):737–55. doi: 10.1177/0333102416651284
36. Fear D, Patel M, Zand R. Serial magnetic resonance imaging findings during severe attacks of familial hemiplegic migraine type 2: a case report. BMC Neurol. (2021) 21(1):173. doi: 10.1186/s12883-021-02201-z
37. Lebas A, Guyant-Maréchal L, Hannequin D, Riant F, Tournier-Lasserve E, Parain D. Severe attacks of familial hemiplegic migraine, childhood epilepsy and ATP1A2 mutation. Cephalalgia. (2008) 28(7):774–7. doi: 10.1111/j.1468-2982.2008.01603.x
38. Costa C, Prontera P, Sarchielli P, Tonelli A, Bassi MT, Cupini LM, et al. A novel ATP1A2 gene mutation in familial hemiplegic migraine and epilepsy. Cephalalgia. (2014) 34(1):68–72. doi: 10.1177/0333102413498941
39. Castro MJ, Nunes B, de Vries B, Lemos C, Vanmolkot KR, van den Heuvel JJ, et al. Two novel functional mutations in the Na+,K+-ATPase alpha2-subunit ATP1A2 gene in patients with familial hemiplegic migraine and associated neurological phenotypes. Clin Genet. (2008) 73(1):37–43. doi: 10.1111/j.1399-0004.2007.00918.x
40. Wang P, Yang YR, Zhang HB, Wang JH, Wang Y. Cognitive dysfunction in a patient with migraine and APT1A2 mutation: a case report. Neurol Sci. (2021) 42(12):5425–31. doi: 10.1007/s10072-021-05284-1
41. Mjåset C, Russell MB. Intravenous nimodipine worsening prolonged attack of familial hemiplegic migraine. J Headache Pain. (2008) 9(6):381–4. doi: 10.1007/s10194-008-0074-2
42. Oda I, Danno D, Saigoh K, Wolf J, Kawashita N, Hirano M, et al. Hemiplegic migraine type 2 with new mutation of the ATP1A2 gene in Japanese cases. Neurosci Res. (2022) 180:83–9. doi: 10.1016/j.neures.2022.03.002
43. Roth C, Freilinger T, Kirovski G, Dunkel J, Shah Y, Wilken B, et al. Clinical spectrum in three families with familial hemiplegic migraine type 2 including a novel mutation in the ATP1A2 gene. Cephalalgia. (2014) 34(3):183–90. doi: 10.1177/0333102413506128
44. Tang W, Zhang M, Qiu E, Kong S, Li Y, Liu H, et al. A Chinese family with familial hemiplegic migraine type 2 due to a novel missense mutation in ATP1A2. Cephalalgia. (2019) 39(11):1382–95. doi: 10.1177/0333102419847738
45. De Cunto A, Bensa M, Tonelli A. A case of familial hemiplegic migraine associated with a novel ATP1A2 gene mutation. Pediatr Neurol. (2012) 47(2):133–6. doi: 10.1016/j.pediatrneurol.2012.04.012
46. Pisano T, Spiller S, Mei D, Guerrini R, Cianchetti C, Friedrich T, et al. Functional characterization of a novel C-terminal ATP1A2 mutation causing hemiplegic migraine and epilepsy. Cephalalgia. (2013) 33(16):1302–10. doi: 10.1177/0333102413495116
47. Antonaci F, Ravaglia S, Grieco GS, Gagliardi S, Cereda C, Costa A. Familial hemiplegic migraine type 2 due to a novel missense mutation in ATP1A2. J Headache Pain. (2021) 22(1):12. doi: 10.1186/s10194-021-01221-x
48. Perrotta A, Gambardella S, Ambrosini A, Anastasio MG, Albano V, Fornai F, et al. A novel ATP1A2 gene variant associated with pure sporadic hemiplegic migraine improved after patent foramen ovale closure: a case report. Front Neurol. (2018) 9:332. doi: 10.3389/fneur.2018.00332
49. Rispoli MG, Di Stefano V, Mantuano E, De Angelis MV. Novel missense mutation in the ATP1A2 gene associated with atypical sporapedic hemiplegic migraine. BMJ Case Rep. (2019) 12(10):e231129. doi: 10.1136/bcr-2019-231129
50. Vanmolkot KR, Kors EE, Hottenga JJ, Terwindt GM, Haan J, Hoefnagels WA, et al. Novel mutations in the Na+, K+-ATPase pump gene ATP1A2 associated with familial hemiplegic migraine and benign familial infantile convulsions. Ann Neurol. (2003) 54(3):360–6. doi: 10.1002/ana.10674
51. Dreier JP, Jurkat-Rott K, Petzold GC, Tomkins O, Klingebiel R, Kopp UA, et al. Opening of the blood-brain barrier preceding cortical edema in a severe attack of FHM type II. Neurology. (2005) 64(12):2145–7. doi: 10.1212/01.Wnl.0000176298.63840.99
52. Vahedi K, Denier C, Ducros A, Bousson V, Levy C, Chabriat H, et al. CACNA1A gene de novo mutation causing hemiplegic migraine, coma, and cerebellar atrophy. Neurology. (2000) 55(7):1040–2. doi: 10.1212/wnl.55.7.1040
53. Zhang L, Wen Y, Zhang Q, Chen Y, Wang J, Shi K, et al. CACNA1A gene variants in eight Chinese patients with a wide range of phenotypes. Front Pediatr. (2020) 8:577544. doi: 10.3389/fped.2020.577544
54. Camia F, Pisciotta L, Morana G, Schiaffino MC, Renna S, Carrera P, et al. Combined early treatment in hemiplegic attacks related to CACNA1A encephalopathy with brain oedema: blocking the cascade? Cephalalgia. (2017) 37(12):1202–6. doi: 10.1177/0333102416668655
55. Debiais S, Hommet C, Bonnaud I, Barthez MA, Rimbaux S, Riant F, et al. The FHM1 mutation S218l: a severe clinical phenotype? A case report and review of the literature. Cephalalgia. (2009) 29(12):1337–9. doi: 10.1111/j.1468-2982.2009.01884.x
56. Di Cristofori A, Fusi L, Gomitoni A, Grampa G, Bersano A. R583q CACNA1A variant in SHM1 and ataxia: case report and literature update. J Headache Pain. (2012) 13(5):419–23. doi: 10.1007/s10194-012-0444-7
57. Beauvais K, Cavé-Riant F, De Barace C, Tardieu M, Tournier-Lasserve E, Furby A. New CACNA1A gene mutation in a case of familial hemiplegic migraine with status epilepticus. Eur Neurol. (2004) 52(1):58–61. doi: 10.1159/000079546
58. Yuan X, Zheng Y, Gao F, Sun W, Wang Z, Zhao G. Case report: a novel CACNA1A mutation caused flunarizine-responsive type 2 episodic ataxia and hemiplegic migraine with abnormal MRI of cerebral white matter. Front Neurol. (2022) 13:899813. doi: 10.3389/fneur.2022.899813
59. Luan H, Zhang L, Zhang S, Zhang M. Next-generation sequencing identified a novel CACNA1A I1379F variant in a familial hemiplegic migraine type 1 pedigree: a case report. Medicine. (2021) 100(51):e28141. doi: 10.1097/md.0000000000028141
60. Sprouse Blum AS, Couperus CJ, Rosen BJ, Haskin-Leahy LF, Shapiro RE. Familial “diplegic” migraine—description of a family with a novel CACNA1A mutation. Headache. (2020) 60(3):600–6. doi: 10.1111/head.13741
61. Carreño O, García-Silva MT, García-Campos Ó, Martínez-de Aragón A, Cormand B, Macaya A. Acute striatal necrosis in hemiplegic migraine with de novo CACNA1A mutation. Headache. (2011) 51(10):1542–6. doi: 10.1111/j.1526-4610.2011.02014.x
62. Pelzer N, Hoogeveen ES, Ferrari MD, Poll-The BT, Kruit MC, Terwindt GM. Brain atrophy following hemiplegic migraine attacks. Cephalalgia. (2018) 38(6):1199–202. doi: 10.1177/0333102417723569
63. Carreño O, Corominas R, Serra SA, Sintas C, Fernández-Castillo N, Vila-Pueyo M, et al. Screening of CACNA1A and ATP1A2 genes in hemiplegic migraine: clinical, genetic, and functional studies. Mol Genet Genomic Med. (2013) 1(4):206–22. doi: 10.1002/mgg3.24
64. Malpas TJ, Riant F, Tournier-Lasserve E, Vahedi K, Neville BG. Sporadic hemiplegic migraine and delayed cerebral oedema after minor head trauma: a novel de novo CACNA1A gene mutation. Dev Med Child Neurol. (2010) 52(1):103–4. doi: 10.1111/j.1469-8749.2009.03493.x
65. Takahashi T, Igarashi S, Kimura T, Hozumi I, Kawachi I, Onodera O, et al. Japanese cases of familial hemiplegic migraine with cerebellar ataxia carrying a T666M mutation in the CACNA1A gene. J Neurol Neurosurg Psychiatry. (2002) 72(5):676–7. doi: 10.1136/jnnp.72.5.676-a
66. Gajam S, Peterson RR, Mathew AA, Thomas A. Sporadic hemiplegic migraine with CACNA1A mutation masquerading as acute meningoencephalitis. Ann Indian Acad Neurol. (2022) 25(3):528–9. doi: 10.4103/aian.aian_908_21
67. Battistini S, Stenirri S, Piatti M, Gelfi C, Righetti PG, Rocchi R, et al. A new CACNA1A gene mutation in acetazolamide-responsive familial hemiplegic migraine and ataxia. Neurology. (1999) 53(1):38–43. doi: 10.1212/wnl.53.1.38
68. Gallanti A, Tonelli A, Cardin V, Bussone G, Bresolin N, Bassi MT. A novel de novo nonsense mutation in ATP1A2 associated with sporadic hemiplegic migraine and epileptic seizures. J Neurol Sci. (2008) 273(1–2):123–6. doi: 10.1016/j.jns.2008.06.006
69. Spadaro M, Ursu S, Lehmann-Horn F, Veneziano L, Antonini G, Giunti P, et al. A G301R Na+/K+-ATPase mutation causes familial hemiplegic migraine type 2 with cerebellar signs. Neurogenetics. (2004) 5(3):177–85. doi: 10.1007/s10048-004-0183-2
70. Vanmolkot KR, Kors EE, Turk U, Turkdogan D, Keyser A, Broos LA, et al. Two de novo mutations in the Na,K-ATPase gene ATP1A2 associated with pure familial hemiplegic migraine. Eur J Hum Genet. (2006) 14(5):555–60. doi: 10.1038/sj.ejhg.5201607
71. Merwick A, Fernandez D, McNamara B, Harrington H. Acute encephalopathy in familial hemiplegic migraine with ATP1A2 mutation. BMJ Case Rep. (2013) 2013:bcr2013009750. doi: 10.1136/bcr-2013-009750
72. Murphy OC, Merwick A, O'Mahony O, Ryan AM, McNamara B. Familial hemiplegic migraine with asymmetric encephalopathy secondary to ATP1A2 mutation: a case series. J Clin Neurophysiol. (2018) 35(1):e3–7. doi: 10.1097/wnp.0000000000000387
73. Du Y, Li C, Duan FJ, Zhao C, Zhang W. Early treatment in acute severe encephalopathy caused by ATP1A2 mutation of familial hemiplegic migraine type 2: case report and literature review. Neuropediatrics. (2020) 51(3):215–20. doi: 10.1055/s-0039-3400986
74. Martínez E, Moreno R, López-Mesonero L, Vidriales I, Ruiz M, Guerrero AL, et al. Familial hemiplegic migraine with severe attacks: a new report with ATP1A2 mutation. Case Rep Neurol Med. (2016) 2016:3464285. doi: 10.1155/2016/3464285
75. Vanmolkot KR, Stam AH, Raman A, Koenderink JB, de Vries B, van den Boogerd EH, et al. First case of compound heterozygosity in Na,K-ATPase gene ATP1A2 in familial hemiplegic migraine. Eur J Hum Genet. (2007) 15(8):884–8. doi: 10.1038/sj.ejhg.5201841
76. Dannenberg F, Prager C, Schmidt F, Tietze A, Bittigau P, Kaindl AM. Intravenous nimodipine treatment for severe episode of ATP1A2 hemiplegic migraine. Pediatr Neurol. (2020) 112:71–2. doi: 10.1016/j.pediatrneurol.2020.07.009
77. Aceves J, Mungall D, Kirmani BF. Sporadic hemiplegic migraine with ATP1A2 and prothrombin gene mutations. Case Rep Neurol Med. (2013) 2013:895057. doi: 10.1155/2013/895057
78. Vanmolkot KR, Stroink H, Koenderink JB, Kors EE, van den Heuvel JJ, van den Boogerd EH, et al. Severe episodic neurological deficits and permanent mental retardation in a child with a novel FHM2 ATP1A2 mutation. Ann Neurol. (2006) 59(2):310–4. doi: 10.1002/ana.20760
79. Li M, Zheng X, Zhong R, Zhao Q, Lu Y, Wang Z, et al. Familial hemiplegic migraine with progressive cerebellar ataxia caused by a p.Thr666Met CACNA1A gene mutation in a Chinese family. Front Neurol. (2019) 10:1221. doi: 10.3389/fneur.2019.01221
80. Stuart S, Roy B, Davies G, Maksemous N, Smith R, Griffiths LR. Detection of a novel mutation in the CACNA1A gene. Twin Res Hum Genet. (2012) 15(1):120–5. doi: 10.1375/twin.15.1.120
81. Toldo I, Cecchin D, Sartori S, Calderone M, Mardari R, Cattelan F, et al. Multimodal neuroimaging in a child with sporadic hemiplegic migraine: a contribution to understanding pathogenesis. Cephalalgia. (2011) 31(6):751–6. doi: 10.1177/0333102410392068
82. Jurkat-Rott K, Freilinger T, Dreier JP, Herzog J, Göbel H, Petzold GC, et al. Variability of familial hemiplegic migraine with novel A1A2 Na+/K+-ATPase variants. Neurology. (2004) 62(10):1857–61. doi: 10.1212/01.wnl.0000127310.11526.fd
83. Segarra NG, Gautschi I, Mittaz-Crettol L, Kallay Zetchi C, Al-Qusairi L, Van Bemmelen MX, et al. Congenital ataxia and hemiplegic migraine with cerebral edema associated with a novel gain of function mutation in the calcium channel CACNA1A. J Neurol Sci. (2014) 342(1–2):69–78. doi: 10.1016/j.jns.2014.04.027
84. Cevoli S, Pierangeli G, Monari L, Valentino ML, Bernardoni P, Mochi M, et al. Familial hemiplegic migraine: clinical features and probable linkage to chromosome 1 in an Italian family. Neurol Sci. (2002) 23(1):7–10. doi: 10.1007/s100720200016
85. Sánchez-Albisua I, Schöning M, Jurkat-Rott K, Lerche H. Possible effect of corticoids on hemiplegic attacks in severe hemiplegic migraine. Pediatr Neurol. (2013) 49(4):286–8. doi: 10.1016/j.pediatrneurol.2013.04.011
Keywords: pediatric, hemiplegic migraine, encephalopathy, diagnosis, treatment
Citation: Xiang Y, Li F, Song Z, Yi Z, Yang C, Xue J and Zhang Y (2023) Two pediatric patients with hemiplegic migraine presenting as acute encephalopathy: case reports and a literature review. Front. Pediatr. 11:1214837. doi: 10.3389/fped.2023.1214837
Received: 30 April 2023; Accepted: 10 July 2023;
Published: 28 July 2023.
Edited by:
Andrea Martinuzzi, Eugenio Medea (IRCCS), ItalyReviewed by:
Alessandro Capuano, Azienda Sanitaria Locale di Viterbo, Italy© 2023 Xiang, Li, Song, Yi, Yang, Xue and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Zhang emhhbmd5aW5nMDEyMjVAcWR1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.