 Beibei Ren1,2,3
Beibei Ren1,2,3 Yu Chen1,2,3Xuanye Bai1,2,3Jiawen Zheng4Jing Chang5Xiangnan Jiang6Qingxin Xia1,2,3,†,‡
Yu Chen1,2,3Xuanye Bai1,2,3Jiawen Zheng4Jing Chang5Xiangnan Jiang6Qingxin Xia1,2,3,†,‡ He Zhang1,2,3,†,‡
He Zhang1,2,3,†,‡
- 1Department of Pathology, Affiliated Cancer Hospital of Zhengzhou University, Zhengzhou, China
- 2Henan Medical Key Laboratory of Tumor Pathology and Artificial Intelligence Diagnosis, Zhengzhou, China
- 3Pathological Diagnostic Antibody Engineering Research Center of Henan Province, Zhengzhou, China
- 4Department of Molecular Pathology, Affiliated Cancer Hospital of Zhengzhou University, Zhengzhou, China
- 5Medical Service Office, Affiliated Cancer Hospital of Zhengzhou University, Zhengzhou, China
- 6Department of Pathology, Fudan University Shanghai Cancer Center, Shanghai, China
Pediatric-type follicular lymphoma (PTFL) is a rare pediatric-type indolent B-cell lymphoma that clinicopathologically differs from adult lymphoma. Accurate diagnosis of PTFL, which is often challenging, is essential to avoid missed diagnosis, misdiagnosis, and overtreatment. To improve our understanding of PTFL, clinicopathological features, differential diagnosis, and molecular mutation characteristics of four patients of PTFL were analyzed using hematoxylin and eosin staining, immunohistochemistry, polymerase chain reaction, fluorescence in situ hybridization (FISH), and next-generation sequencing (NGS). A relevant literature review was also performed. All four PTFL patients were male, with ages of 6, 18, 13, and 15 years, and had St. Jude stage I or III. Microscopic results showed that the structure of the lymph nodes was destroyed; the tumor follicles were enlarged and irregular; medium–large blastoid cells with a consistent shape were visible in tumor follicles, and the nucleus was round or oval; and the “starry sky” pattern was easily observed. Tumor cells expressed CD20, PAX-5, BCL6, and CD10. None of the tumor cells expressed BCL2, CD3, CD5, MUM1, and CyclinD1. CD21 showed dilated growth of a follicular dendritic cell network in tumor follicles. EBER genes were negative in all cases. FISH testing also showed negative BCL2 gene breaks and IRF4 gene breaks in all cases. NGS detected 12 related mutant genes, including KMT2D, CD79B, GNA13, MYD88, PCLO, TCF3, IRF8, MAP2K1, FOXO1, POLE, INPP5D, and FAT4. Two of the four patients had an IRF8 gene mutation, and one patient had a dual mutation of the MAP2K1 gene. Our study revealed the unique clinicopathological features and molecular mutational characteristics of PTFL, consolidated our understanding of PTFL, and identified other rare mutant genes, which may further contribute to the study of the molecular mechanism and differential diagnosis of PTFL.
1. Introduction
Follicular lymphoma (FL) is a mature lymphoid malignancy derived from germinal center B cells, accounting for approximately 22% of all non-Hodgkin's lymphomas. It is common in middle-aged and older people but rare in children and young people. In 2008, the World Health Organization (WHO) classification of lymphoid tissue and hematopoietic system first proposed “pediatric follicular lymphoma” (1). Because of its unique clinical pathology and molecular genetic characteristics, pediatric-type follicular lymphoma (PTFL) was recognized as a distinct entity in the revised 4th edition of the WHO classification of lymphoid neoplasms and was discussed separately in the 5th edition of the WHO classification of lymphoma (2, 3).
The development and maturity of next-generation sequencing (NGS) technology have provided a good platform for the in-depth study of the molecular mechanism associated with the occurrence, development, and prognosis of FL. However, research on NGS-based gene mutations in PTFL is currently limited. In this report, data from four PTFL cases were obtained from the Clinical Pathology Center of the Affiliated Cancer Hospital of Zhengzhou University and the Department of Pathology of Shanghai Cancer Center of Fudan University, and clinicopathological and molecular mutation characteristics were analyzed to improve our understanding of PTFL.
2. Materials and methods
2.1. Case description
Data from four patients of PTFL were obtained from the Clinical Pathology Center of the Affiliated Cancer Hospital of Zhengzhou University and the Department of Pathology of Shanghai Cancer Center of Fudan University between June 2019 and August 2021. Two pathologists blinded to the molecular results reviewed the cases, and a consensus diagnosis was achieved based on the revised WHO classification of lymphoma (2021 version). One of the specimens was obtained by needle biopsy, and the other two were obtained through excisional biopsy. Relevant clinical and survival data were obtained using the electronic medical record system and through a telephone follow-up. This study was approved by the Institutional Review Boards of the two hospitals and was conducted according to the Declaration of Helsinki.
2.2. Hematoxylin and eosin (H&E) and immunohistochemistry (IHC) staining
Tissues were fixed in a 3.7% neutral formaldehyde solution, dehydrated, embedded in paraffin, and sectioned at 4 μm thickness. H&E staining was used to evaluate lymph node biopsies. IHC staining was performed for CD20, CD3, CD5, CD10, BCL2, BCL6, MUM1, CD21, CyclinD1, and Ki-67 as part of the diagnostic workup. Immunostaining was performed on Ventana BenchMark ULTRA following the protocol of the manufacturer. CD20, CD21, CD3, CD5, and CD10 were located in the cell membrane; BCL2 was located in the cell membrane/cytoplasm; and MUM1, BCL6, CyclinD1, and Ki-67 were located in the nucleus.
2.3. Epstein–Barr virus–encoded small RNA (EBER) and fluorescence in situ hybridization (FISH)
The expression of EBER in PTFL paraffin sections was also detected using Ventana BenchMark ULTRA. Epstein–Barr virus (EBV) in situ hybridization was performed using the EBER probe, which was purchased from Roche Diagnostics (Shanghai) Co., Ltd. (Shanghai, China). The experimental procedures were performed according to the instructions of the manufacturer. The nucleus was stained dark blue (positive signal), while the cytoplasm was stained light red. EBV-positive nasopharyngeal carcinoma was selected as positive control. In addition, the BCL2 and IRF4 gene rearrangements were detected using fluorescence in situ hybridization (FISH) assays using commercial reagent kits (BCL2 and IRF4 Dual Color Break Apart Rearrangement probe, Anbiping Co., Ltd.). A double-color breakage probe contains two probes labeled with green and red fluorescence, respectively. In cells without gene breakage, a yellow fusion signal appears (red and green signals overlap to form a yellow signal), or red and green signals adhere to each other (the interval is less than two signal diameters). In broken genes, a fused yellow signal, a separate green signal, and a separate red signal appear in the nucleus.
2.4. B-cell clonality test
Genomic DNA was extracted from formalin-fixed, paraffin-embedded (FFPE) tissue sections using the Maxwell 16 FFPE Tissue LEV DNA Purification Kit and the Maxwell 16 Instrument, following the instructions of the manufacturer. Polymerase chain reaction amplification was performed to detect clonal immunoglobulin (Ig) heavy and/or Ig kappa light chain gene rearrangements, following the BIOMED-2 protocol. The criteria for defining a positive band are as follows: Products generated from diagnostic samples that fall within the valid size range and are at least three times the amplitude of the third largest peak in the polyclonal background are consistent with a positive peak. One or two prominent positive bands within the valid size range are reported as “positive for the detection of clonal immunoglobulin heavy chain or kappa light chain gene rearrangement(s) consistent with the presence of a clonal cell population. In the context of overall diagnostic criteria, clonal cell populations can indicate the presence of hematologic malignancy.” In contrast, an absence of positive bands within the valid size range is reported as “negative for the detection of clonal immunoglobulin heavy chain or kappa light chain gene rearrangement(s).”
2.5. Targeted NGS analysis
Targeted NGS was performed on the FFPE tissue samples of the patients using a customized panel of 93 lymphoma-related genes, related to auxiliary diagnosis, prognostic assessment, and targeted drug use of lymphoma. Genomic DNA was extracted from FFPE tissues using the Gene+OncoLym FFPE Kit. Single-nucleotide variants (SNVs) and small insertions and deletions (indels) were detected using an in-house developed pipeline, SNVer and LoFreq. Translocations were detected using Delly and Mantana. Mutations were annotated using SnpEff.
3. Results
3.1. Clinical characteristics
Case 1 involves a 6-year-old male patient who presented to our hospital for the evaluation of a non-tender mass over the right neck area. A positron emission tomography–computed tomography (CT) scan showed multiple soft tissue nodule shadows in the right cervical regions Ⅱ and Ⅲ with increased metabolism, indicating the presence of malignant lesions of multiple lymph nodes. A biopsy of the right neck was performed for pathological diagnosis. No operation or chemotherapy was performed after the diagnosis using a needle biopsy. The patient underwent follow-up for 28 months without disease progression (Table 1).
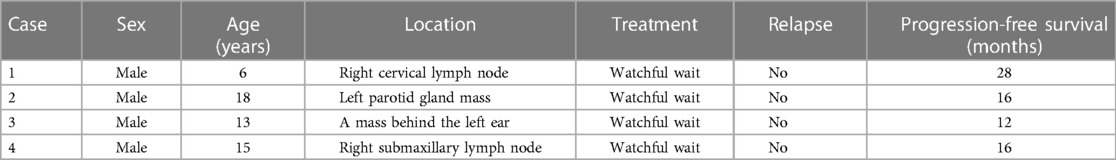
Table 1. Clinical characteristics of the four patients with PTFL.
Case 2 involves an 18-year-old male patient who presented to our hospital for the evaluation of a slowly growing and non-tender mass over the left parotid gland area. No systemic symptoms, such as fever, night sweats, and weight loss, were present. A preoperative ultrasound showed a solid mass in the left parotid gland but no abnormal lymphadenopathy in the bilateral neck. There was a solid space-occupying lesion but no abnormal enlargement of lymph nodes. Mass removal with superficial parotidectomy was performed with routine precautions to preserve the facial nerve and the parotid duct. No further treatment following the simple surgical resection was performed. The patient underwent follow-up for 16 months and survived without a tumor (Table 1).
Case 3 involves a 13-year-old male patient who presented with a mass behind the left ear area that had been present for 3 months. No systemic symptoms, such as fever, night sweats, and weight loss, were present. After surgical resection, the patient underwent follow-up for 12 months and survived without a tumor (Table 1).
Case 4 involves a 15-year-old male patient who presented with a non-tender mass on the right neck area that had been present for 2 months. A preoperative CT scan showed that the lymph nodes in the right submaxillary region were swollen, and there were numerous small lymph nodes in the two necks, some of which were slightly larger. After surgical resection, the patient underwent follow-up for 16 months and survived without a tumor (Table 1).
3.2. Morphological findings
Taking Case 2 as an example, H&E staining showed extensive proliferation of follicular cells. Under a low-power microscope, it was observed that the structure of the lymph nodes was destroyed, and crowded, swollen, and irregular follicle-like structures with different sizes were seen, which were creeping forms (Figure 1A). The “starry sky” pattern was easily observed, normally a feature of reactive follicles caused by tangible body macrophages. The polar distribution of the light and dark areas of the follicles disappeared, and the mantle area became thinner or disappeared in some areas (Figure 1B). At high magnification, neoplastic follicles were found to be composed of uniform, medium-to-large blastoid cells (Figure 1C). In addition, there is no histologic heterogeneity throughout the lymph node. Follicle centers were strongly positive for BCL6 and CD20 (Figure 1D) and negative for BCL2 (Figure 1G), CD3 (Figure 1E), CD5, and CyclinD1. Neoplastic follicles of two cases were positive for CD10 (Figure 1F), and one case was positive for MUM1 (Figure 1H). CD21 showed dilated growth of a follicular dendritic cell network in the neoplastic follicles. In addition, Ki-67 was highly expressed in all four cases (Figure 1I).
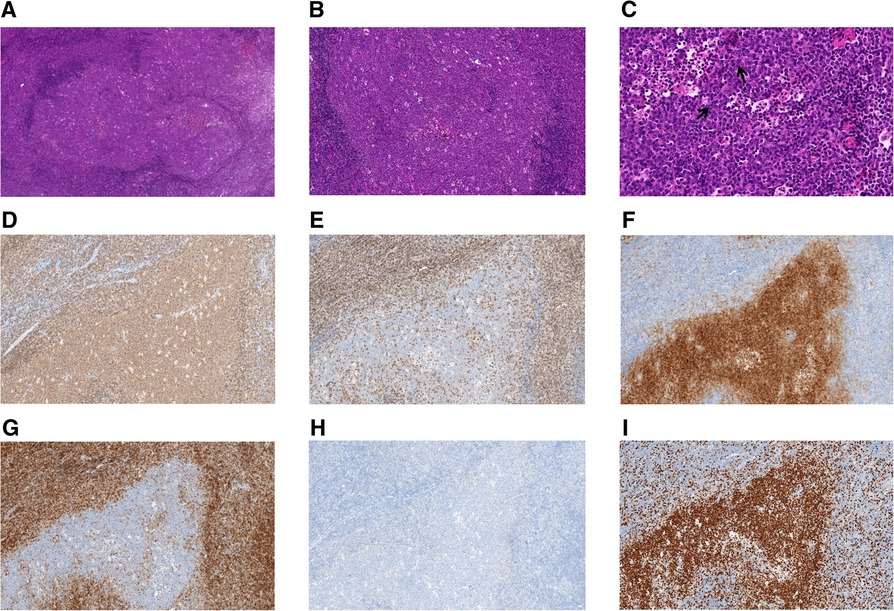
Figure 1. Histologic and immunohistochemical features of pediatric-type follicular lymphoma (Case 2). (A) Representative images of hematoxylin and eosin staining. Neoplastic follicles are enlarged, irregular, and creeping, and their mantles are thinned (original magnification, 40×). (B) Numerous macrophages with tangible bodies confer a starry sky appearance (original magnification, 100×). (C) Lymphoma cells are monotonous, intermediate-sized, and morphologically different from centrocytes or centroblasts in adult FL. The cells are typically blastic in appearance, with dispersed chromatin and inconspicuous nucleoli (original magnification, 400×). ↑: blastoid cells. (D–I) Immunohistochemistry of PTFL EnVision (original magnification, 100×). (D) CD20, (E) CD3, (F) CD10, (G) BCL2, (H) MUM1, and (I) Ki-67.
3.3. FISH and B-cell clonality test
EBER hybridization, BCL2 gene breaks, and IRF4 gene breaks were all negative in the four cases (Figures 2A–C), while the IG gene rearrangements were all positive (Figure 2D). There were polyclonal backgrounds, but no oligoclonal backgrounds, in the four cases. Products generated from the four diagnostic samples that fall within the valid size range and are at least three times the amplitude of the third largest peak in the polyclonal background are consistent with a positive peak; therefore, the detection of clonal immunoglobulin gene rearrangements was positive.
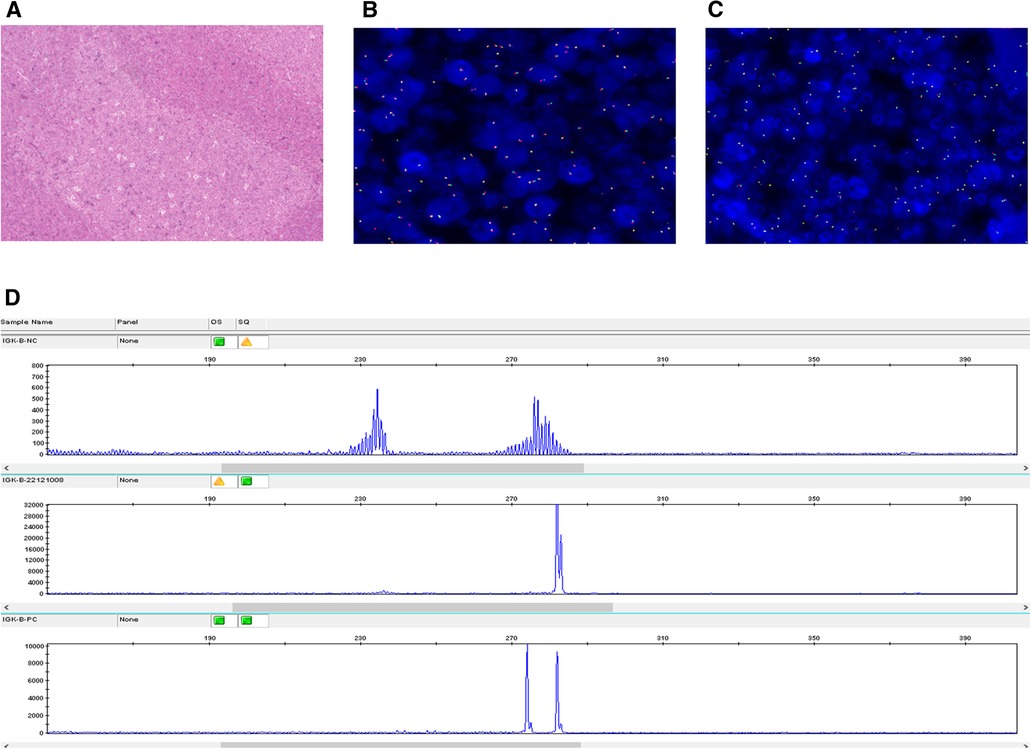
Figure 2. EBER, BCL-2, and B-cell monoclonal rearrangement analysis (Case 2). (A) EBER hybridization was negative. (B) FISH was negative for BCL2 rearrangements. (C) FISH was negative for IRF4 rearrangements. (D) IGK gene rearrangement was positive.
3.4. Mutational landscape
Targeted NGS was performed on all four cases. A total of 15 SNVs or indels were detected in 12 genes, including PCLO, CD79B, MYD88, GNA13, KMT2D, TCF3, MAP2K1, IRF8, FOXO1, POLE, INPP5D, and FAT4. Two of the four cases had IRF8 gene mutation (Table 2), and one case had a dual mutation of the MAP2K1 gene.
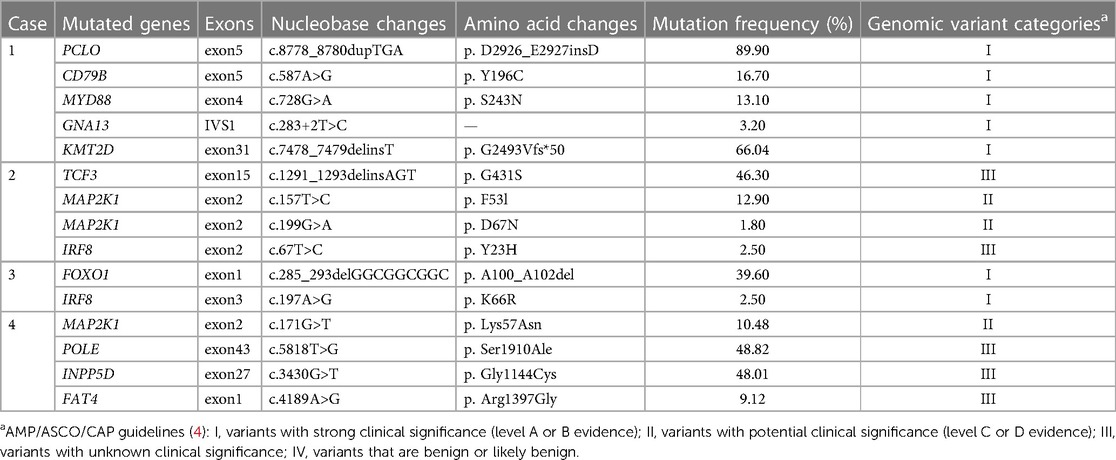
Table 2. Genetic alterations of the four PTFL patients.
4. Discussion
In 1979, Frizzera et al. (5) proposed for the first time that FL in children had unique clinical and morphological characteristics. Subsequently, several studies found that FL in children differs from typical FL in adults in terms of histomorphology, molecular genetics, immunophenotyping, and prognosis. Thus, PTFL was officially recognized as a definitive entity in the updated 2016 WHO classification. PTFL is a rare indolent B-cell lymphoma (2). In this study, we retrospectively analyzed the clinicopathological and genetic features of four PTFL cases and reviewed related pieces of literature.
Although PTFL was initially identified in children, it was subsequently found in young adults and even older patients (6). PTFL exhibits a predominance in men, with a male-to-female ratio ranging from approximately 4:1 to 10:1 (7). The four patients in our study were 6, 18, 13, and 15 years old, respectively, and all were males. PTFL most commonly occurs in the lymph nodes in the head and neck but is less frequent in the inguinal and axillary nodes. The tonsil often occurs outside the node. PTFL can also arise in the liver, spleen, mediastinum, parotid gland, and other parts (8, 9). Our patients presented with enlargement of the right cervical lymph node, left parotid gland mass, and left ear mass. PTFL in the parotid gland area of children is rare. Jangyoun et al. reported one case of PTFL occurring in the left parotid gland region (9), which had similar clinical characteristics to Case 2 in our study. PTFL is a localized disease, often without systemic symptoms; most cases are diagnosed at stage I or II, which can be treated by simple surgical resection. Occasionally, chemotherapy is a feasible option in patients with slightly advanced PTFL that is difficult to resect surgically, with a good prognosis (6, 10). Case 1 was diagnosed with clinical stage III and underwent follow-up for 28 months. No operation or chemotherapy was performed. Case 2 was diagnosed with clinical stage I and underwent left parotidectomy and follow-up for 16 months. Case 3 was diagnosed with clinical stage I and underwent simple resection and follow-up for 12 months. Case 4 was diagnosed with clinical stage I and underwent simple resection and follow-up for 16 months. No disease progression was observed in all cases.
Histopathologically, the lymph node exhibits an effaced architecture and large expansile follicles with attenuated mantle zones and a serpiginous growth pattern. The follicles are non-polarized, but a partial starry sky pattern with tangible body macrophages may be observed. Lymphoma cells are monotonous, intermediate-sized, and morphologically different from centrocytes or centroblasts in adult FL. Lymphoma cells are typically blastic in appearance, with dispersed chromatin and inconspicuous nucleoli. Immunophenotypically, lymphoma cells express germinal centripetal markers such as CD10, BCL6, and HGAL and mature B-cell markers such as CD20, CD79a, and PAX-5 (3, 11). Most cases exhibit a high Ki-67 proliferation index, but not MUM1, CyclinD1, and C-myc. The BCL2 protein is often negative, but a minority of patients present a weak intensity. Neither t (4;18) translocation nor BCL2, BCL6, MYC, and IRF4 rearrangements were observed (12, 13). Nevertheless, all PTFL were positive for the IG gene rearrangement. Interestingly, the clinical features, pathological morphology, immunophenotype, and molecular genetics of the four patients in this study were consistent with the above characteristics.
With the development of high-throughput sequencing, studies have disclosed that BCL2 rearrangement and mutations in histone methyltransferases (KMT2D and EZH2) and acetyltransferases (CREBBP and EP300) are the hallmarks of FL (12, 14). However, low genomic complexity and frequent aberrations in TNFRSF14, MAP2K1, and IRF8 genes characterize PTFL (12, 15, 16). It lacks the BCL2 gene rearrangement, and mutations in epigenetic modifier genes, including KMT2D, CREBBP, and EP300, are less common. Our study detected 15 mutations in 12 genes. Two cases exhibited IRF8 gene mutation, and none of the four cases had TNFRSF14 mutation. According to the guidelines for somatic cell variation interpretation jointly formulated by the American Society for Clinical Pathology (AMP)/American Society of Clinical Oncology (ASCO)/College of American Pathologists (CAP) in 2017, all gene variations in Cases 1 and 3 were of grade I, which has a strong clinical significance in cancer diagnosis, prognosis, and/or therapeutics. CD79B, MYD88, and IRF8 were missense mutations. PCLO and KMT2D were insertion mutations, of which the mutation frequency of PCLO was quite high. FOXO1 was a deletion mutation, while GNA13 was a splicing mutation in intron 1, leading to abnormal splicing of messenger RNA, thus affecting protein function. The genetic variations in Cases 2 and 4 were of grade III, except for MAP2K1, which was classified as grade II. The clinical significance of grade III variants was unclear, while grade II variants (with evidence of Class C or D) had potential clinical significance. In Cases 2 and 4, the MAP2K1 mutation was repeated within exon 2, which encodes the negative regulatory region domain of the MEK1 protein. TCF3 were insertion mutations. POLE, INPP5D, and FAT4 were missense mutations. At present, there is scarcely any definite conclusion about the mechanism of these rare gene aberrations involved in PTFL, and thus, more prospective studies are needed.
The diagnosis of PTFL is more difficult in clinical practice; therefore, it can easily result in leak diagnosis or misdiagnosis. In addition, the treatment and prognosis of PTFL are quite different from other lymphomas; hence, it should be distinguished from the following diseases:
• Classical FL: it mainly affects adults but rarely occurs in pediatric and young adult populations. Most of the histological grades (approximately 80%) are low grades. The immunophenotypes of FL often express BCL2, BCL6, and CD10. BCL2 and BCL6 gene rearrangements characterize the genetics of FL. In addition, FL in adults with negative BCL2 translocation often expresses CD23 and has a high proliferation rate; however, it lacks CD10 expression, which can be differentiated from PTFL (17).
• IRF4-rearranged large B-cell lymphoma: like PTFL, it occurs in children and young adults. The head and neck regions, such as Waldeyer's ring and cervical lymph nodes, are the most involved. However, the morphology is characterized by a follicular proliferation of medium-sized blastoid cells without BCL2 translocations and a lack of starry sky phenomena. Tumor cells consistently express IRF4/MUM1 with IRF4 gene rearrangements (7).
• Pediatric nodal marginal zone lymphoma (PNMZL): it was included as a provisional entity in the revised 4th WHO classification of lymphoid neoplasms. Morphologically, PNMZL often demonstrates large, expanded follicles disrupted by mantle zone cells, resembling progressive transformation of germinal centers. The atypical B cells in PNMZL often co-express CD43 and may be positive for BCL2, while germinal center B-cell markers (CD10 and BCL6) are often negative. Lymphoma cells are negative for Gide, which highlights the expanded mantle zone B cells in PTGC-like follicles. The Ig gene is clonally rearranged. Recent studies have shown that PNMZL and PTFL show overlapping clinicopathological and molecular genetic characteristics. Julia et al. proposed to rename PNMZL as “PTFL with or without marginal zone differentiation” (18).
• Reactive follicular hyperplasia (RFH): the key architectural findings distinguishing RFH from PTFL (lack of nodal effacement, intact polarization of follicles) are often absent based on cytological studies. Lymph nodes with RFH also contain numerous tangible body macrophages in the background, imparting a starry sky appearance, and the immunophenotype is almost consistent with PTFL (19). Recently, Agostinelli et al. (20) showed that the positive rate of FOXP-1 in PTFL was 92%, but it was negative in the reactive germinal center. This discovery led to the emergence of FOXP-1 as a new diagnostic marker for PTFL and helped distinguish RFH.
As with treating PTFL, the United States National Comprehensive Cancer Network (NCCN) guidelines mainly recommend the watch-and-wait strategy for benign tumors. Chemotherapy is no longer the first-line therapy. However, NCCN still recommends radiotherapy or chemotherapy of the affected regions (when the local disease is severe) as an alternative treatment option (21). Overall, PTFL has a good prognosis, with a 5-year survival rate of >95% and a 2-year disease-free survival rate of approximately 94% after chemotherapy for patients with advanced-stage PTFL (22, 23). In conclusion, studying the clinicopathological and genetic mutation landscape of PTFL can expand our understanding of PTFL, thus avoiding leak diagnosis, misdiagnosis, and overtreatment.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Materials; further inquiries can be directed to the corresponding authors.
Ethics statement
Written informed consent for the publication of any potentially identifiable images or data included in this article was obtained from the individual(s)’ and minor(s)’ legal guardian/next of kin.
Author contributions
(1) Conception and design: HZ. (2) Administrative support: HZ, JC, and QX. (3) Provision of study materials or patients: YC, JZ, XB, and XJ. (4) Collection and assembly of data: BR and XB. (5) Data analysis and interpretation: BR, YC and XB. (6) Manuscript writing: BR. (7) Final approval of manuscript: all authors. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the National Natural Science Foundation of China (Grant Number: 31901233), Medical Science and Technology Joint Construction Project of Henan Province (Grant Number: LHGJ20210174), and Scientific and Technological Breakthroughs in Henan Province (Grant Number: 222102310157).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Jaffe ES. The 2008 who classification of lymphomas: implications for clinical practice and translational research. Hematol Am Soc Hematol Educ Program. (2009) 2009(1):523–31. doi: 10.1182/asheducation-2009.1.523
2. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. (2016) 127(20):2375–90. doi: 10.1182/blood-2016-01-643569
3. Ferry JA. Update from the 5th edition of the World Health Organization classification of head and neck tumors: hematolymphoid proliferations and neoplasia. Head Neck Pathol. (2022) 16(1):101–9. doi: 10.1007/s12105-022-01411-2
4. Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. (2017) 19(1):4–23. doi: 10.1016/j.jmoldx.2016.10.002
5. Frizzera G, Murphy SB. Follicular (nodular) lymphoma in childhood: a rare clinical-pathological entity. Report of eight cases from four cancer centers. Cancer. (1979) 44(6):2218–35. doi: 10.1002/1097-0142(197912)44:6%3C2218::aid-cncr2820440634%3E3.0.co;2-d
6. Oschlies I, Salaverria I, Mahn F, Meinhardt A, Zimmermann M, Woessmann W, et al. Pediatric follicular lymphoma–a clinico-pathological study of a population-based series of patients treated within the non-Hodgkin’s lymphoma–Berlin-Frankfurt-Munster (NHL-BFM) multicenter trials. Haematologica. (2010) 95(2):253–9. doi: 10.3324/haematol.2009.013177
7. Quintanilla-Martinez L, Sander B, Chan JK, Xerri L, Ott G, Campo E, et al. Indolent lymphomas in the pediatric population: follicular lymphoma, IRF4/MUM1+ lymphoma, nodal marginal zone lymphoma and chronic lymphocytic leukemia. Virchows Arch. (2016) 468(2):141–57. doi: 10.1007/s00428-015-1855-z
8. Kobayashi R, Tanaka F, Nakazawa A, Ueyama JI, Sunami S, Mitsui T, et al. Pediatric follicular lymphoma in Japan. Int J Hematol. (2017) 105(6):849–53. doi: 10.1007/s12185-017-2209-1
9. Choi J, Choi HJ, Yim K, Kwon H, Byeon JH, Jung SN. Pediatric follicular lymphoma of the parotid gland. Arch Craniofac Surg. (2018) 19(4):279–82. doi: 10.7181/acfs.2018.02075
10. Xiao-yin P, Wen-cai L, Guan-nan W, Dan-dan Z, Yan-ping Z, Wu-gan Z. Clinicopathological and molecular genetic analysis of 9 cases of paediatric-type follicular lymphoma. J Clin Exp Pathol. (2020) 6(36):662–5. doi: 10.13315/j.cnki.cjcep.2020.06.007
11. Louissaint A Jr, Ackerman AM, Dias-Santagata D, Ferry JA, Hochberg EP, Huang MS, et al. Pediatric-type nodal follicular lymphoma: an indolent clonal proliferation in children and adults with high proliferation index and no BCL2 rearrangement. Blood. (2012) 120(12):2395–404. doi: 10.1182/blood-2012-05-429514
12. Louissaint A Jr, Schafernak KT, Geyer JT, Kovach AE, Ghandi M, Gratzinger D, et al. Pediatric-type nodal follicular lymphoma: a biologically distinct lymphoma with frequent MAPK pathway mutations. Blood (2016) 128(8):1093–100. doi: 10.1182/blood-2015-12-682591
13. Schmidt J, Ramis-Zaldivar JE, Nadeu F, Gonzalez-Farre B, Navarro A, Egan C, et al. Mutations of MAP2K1 are frequent in pediatric-type follicular lymphoma and result in ERK pathway activation. Blood. (2017) 130(3):323–7. doi: 10.1182/blood-2017-03-776278
14. Green MR. Chromatin modifying gene mutations in follicular lymphoma. Blood. (2018) 131(6):595–604. doi: 10.1182/blood-2017-08-737361
15. Schmidt J, Gong S, Marafioti T, Mankel B, Gonzalez-Farre B, Balague O, et al. Genome-wide analysis of pediatric-type follicular lymphoma reveals low genetic complexity and recurrent alterations of Tnfrsf14 gene. Blood. (2016) 128(8):1101–11. doi: 10.1182/blood-2016-03-703819
16. Ozawa MG, Bhaduri A, Chisholm KM, Baker SA, Ma L, Zehnder JL, et al. A study of the mutational landscape of pediatric-type follicular lymphoma and pediatric nodal marginal zone lymphoma. Mod Pathol. (2016) 29(10):1212–20. doi: 10.1038/modpathol.2016.102
17. Jacobsen E. Follicular lymphoma: 2023 update on diagnosis and management. Am J Hematol. (2022) 97(12):1638–51. doi: 10.1002/ajh.26737
18. Salmeron-Villalobos J, Egan C, Borgmann V, Muller I, Gonzalez-Farre B, Ramis-Zaldivar JE, et al. A unifying hypothesis for PNMZL and PTFL: morphological variants with a common molecular profile. Blood Adv. (2022) 6(16):4661–74. doi: 10.1182/bloodadvances.2022007322
19. Lu KL, Menke JR, Ng D, Ruiz-Cordero R, Marinoff A, Stieglitz E, et al. Cytomorphologic features of pediatric-type follicular lymphoma on fine needle aspiration biopsy: case series and a review of the literature. J Am Soc Cytopathol. (2022) 11(5):281–94. doi: 10.1016/j.jasc.2022.06.004
20. Agostinelli C, Akarca AU, Ramsay A, Rizvi H, Rodriguez-Justo M, Pomplun S, et al. Novel markers in pediatric-type follicular lymphoma. Virchows Arch. (2019) 475(6):771–9. doi: 10.1007/s00428-019-02681-y
21. Swerdlow SH, Campo E, Harris NL. WHO classicalion of tumours of haematopoietic and lymphoid tssues. 4th ed. Lyon: IARC Press (2017). p. 280–1.
22. Attarbaschi A, Beishuizen A, Mann G, Rosolen A, Mori T, Uyttebroeck A, et al. Children and adolescents with follicular lymphoma have an excellent prognosis with either limited chemotherapy or with a “watch and wait” strategy after complete resection. Ann Hematol. (2013) 92(11):1537–41. doi: 10.1007/s00277-013-1785-2
Keywords: follicular lymphoma, pediatric type follicular lymphoma, Immunohistochemistry, NGS, molecular
Citation: Ren B, Chen Y, Bai X, Zheng J, Chang J, Jiang X, Xia Q and Zhang H (2023) Case report: Clinicopathological and molecular characteristics of pediatric-type follicular lymphoma. Front. Pediatr. 11:1205384. doi: 10.3389/fped.2023.1205384
Received: 13 April 2023; Accepted: 30 June 2023;
Published: 19 July 2023.
Edited by:
Nicoleta Arva, Northwestern University, United StatesReviewed by:
Alexandra Kovach, Children’s Hospital of Los Angeles, United StatesShunyou Gong, Ann & Robert H. Lurie Children’s Hospital of Chicago, United States
© 2023 Ren, Chen, Bai, Zheng, Chang, Jiang, Xia and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
†These authors have contributed equally to this work and share last authorship
‡ORCID Qingxin Xia orcid.org/0000-0002-0840-9658 He Zhang orcid.org/0000-0001-7729-739X