 Kaiyan Wei
Kaiyan Wei Chaochun Zou*
Chaochun Zou*
- Department of Endocrinology, Children's Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, Hangzhou, China
The NAA10 gene encodes the catalytic subunit of the N-terminal acetyltransferase protein complex A (NatA), which is supposed to acetylate approximately 40% of the human proteins. After the advent of next-generation sequencing, more variants in the NAA10 gene and Ogden syndrome (OMIM# 300855) have been reported. Individuals with NAA10-related syndrome have a wide spectrum of clinical manifestations and the genotype–phenotype correlation is still far from being confirmed. Here, we report a three years old Chinese girl carrying a heterozygous de novo NAA10 [NM_003491: c. 247C > T, p. (Arg83Cys)] variant (dbSNP# rs387906701) (ClinVar# 208664) (OMIM# 300013.0010). The proband not only has some mild and common clinical manifestations, including dysmorphic features, developmental delay, obstructive hypertrophic cardiomyopathy, and arrhythmia, but also shows some rare clinical features such as exophthalmos, blue sclera, cutaneous capillary malformations, and adenoid hypertrophy. Our attempt is to expand the clinical phenotype associated with NAA10-related syndrome and explore genotype–phenotype correlation with such syndrome.
Introduction
N-terminal acetylation (NTA), which is carried out by N-terminal acetyltransferases, is one of the most common protein modifications (1, 2). The N-terminal acetyltransferase protein complex A (NatA)is responsible for N-terminal acetylating approximately 40% of all human proteins and is composed of the catalytic subunit NAA10 and the auxiliary subunits NAA15, NAA50, and HYPK (3, 4). The human NAA10 gene, which encodes the catalytic subunit of the NatA, is an essential gene located in Xq28. Previous studies have shown that many hereditary or de novo NAA10 variants are pathogenic (5–7). Originally, a missense variant NAA10 p. (Ser37Pro) was identified as the reason for an X-linked recessive lethal disorder in eight males from two families, which was called Ogden syndrome (OMIM# 300855) in 2011 (8). With the development of next-generation sequencing technology, a series of new NAA10 variants have been reported, and the phenotypic spectrum of patients has broadened rapidly (6, 9–12). Although postnatal developmental retardation, intellectual disability (ID), and cardiac anomalies may be the main clinical manifestations, individuals exhibit heterogenous phenotypes with no clear genotype-phenotype correlation (5). Therefore, researchers proposed that this series of disorders should be referred to more broadly as NAA10-related syndrome (13).
Here, we report a heterozygous de novo variant of NAA10 in a three-year-old Chinese girl whose whole exome sequencing (WES) identified a mutation of c. 247C > T, p. (Arg83Cys) (dbSNP# rs387906701) (ClinVar# 208664) (OMIM# 300013.0010). The girl showed developmental retardation, intellectual disability, and growth failure, which are common in NAA10-related syndrome. Meanwhile, she did exhibit some special clinical features and a less severe phenotype of cardiovascular defects, as well as recurrent respiratory tract infections. Our report further expands the mutation and clinical spectrum associated with NAA10-related syndrome and provides insight into its natural history and life trajectory, which contributes to the identification and comprehensive study of NAA10-related syndrome.
Case presentation
Our patient was a girl who was the second child of healthy and non-consanguineous parents with Chinese origin. She was born naturally at 40 weeks of gestation. Her global exhibition was normal at birth, and her body weight and body length were 3,960 g and 50 cm, respectively. However, when she was 3 months old, the auscultation of the heart found a grade III murmur of puffing character. Then she was diagnosed with Hypertrophic Cardiomyopathy (HCM) by echocardiography.
Several dysmorphic features were also found in the subsequent physical examination, including thick eyebrows, blue sclera, ocular hypotelorism, prominent eyes, large and low-set ears, broad and flat nasal bridge, flared nares, short columella, wide philtrum and mouth and protruding upper lip (Figures 1A,B). The skin was characterized by redundancy or laxity with cutaneous capillary malformations. While the developmental defects were not present in the extremities.
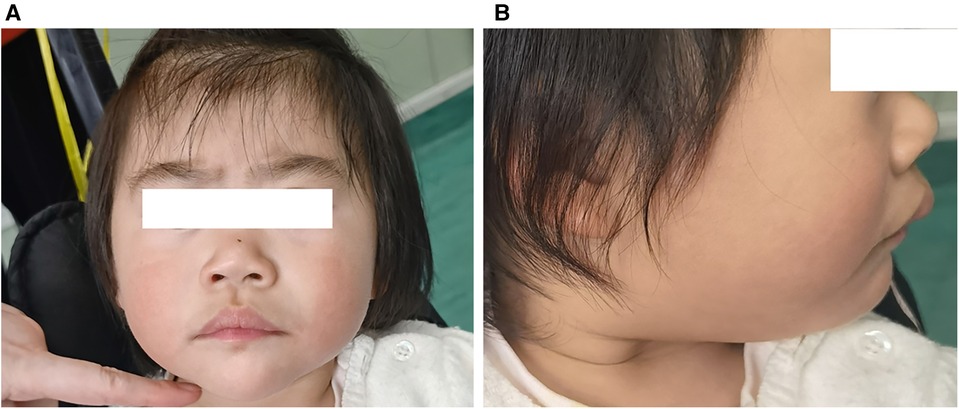
Figure 1. Clinical features of the patient at the age of 2 years: thick eyebrows, broad and flat nasal bridge, flared nares, short columella, wide philtrum, and protruding upper lip (A), large and low-set ears (B).
The echocardiography (ECG) revealed ventricular septal hypertrophy, left ventricular hypertrophy, and left ventricular outflow tract obstruction. The dynamic electrocardiogram (DCG) demonstrated sinus arrhythmia, sporadic premature atrial beats, and frequent premature ventricular beats. The cardiac color ultrasound identified the congenital heart disease with obstructive hypertrophic cardiomyopathy and left ventricular outflow tract stenosis. In addition, the cardiac magnetic resonance imaging (MRI) also showed obstructive hypertrophic cardiomyopathy with heterogeneous left ventricular hypertrophy and left ventricular outflow tract jet sign (Figures 2A–D). The brain MRI examination showed the left pretemporal space was slightly wider. Moreover, the thyroid ultrasonography (USG) examination showed a small thyroid gland, while the thyroid hormone was normal. The chest x-ray increased bronchovascular shadows. While, the visual acuity, hearing tests, and abdominal ultrasonography were normal.
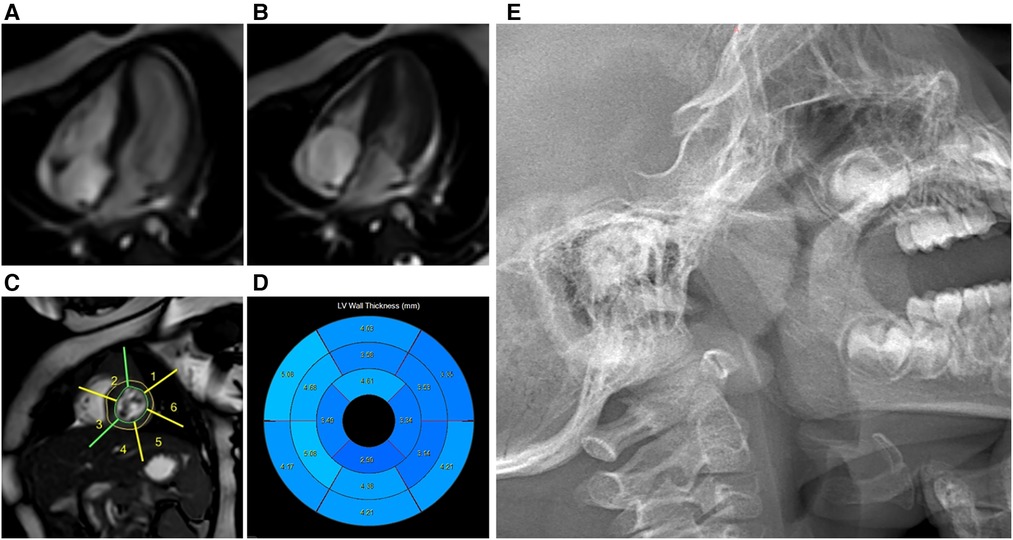
Figure 2. Cardiac MRI: obstructive hypertrophic cardiomyopathy with heterogeneous left ventricular hypertrophy. Cardiac magnetic resonance cine images from mid-ventricular short-axis and four-chamber views at end-diastole (A) and end-systole (B); Cardiac magnetic resonance cine images (C); Bulls-eye plot representing the thickness of the left ventricle (D); Lateral nasopharyngeal x-ray: adenoidal hypertrophy and tonsil hypertrophy (E).
Given the special clinical signs she had presented, whole exome sequencing (WES) was performed when she was 3 months old. The result showed a heterozygous X-linked missense variant in the NAA10 gene [NM_003491: exon5, c. 247C > T, p. (Arg83Cys)], which had been previously reported as pathogenic in variant databases (Clin Var, HGMD) and occurred in a region conserved across species. The Sanger sequencing revealed this variant was not detected in either of her parents, thus arising de novo in the proband. In addition, her karyotype was 46, XX.
Then she had since been treated with metoprolol tartrate tablets to control her arrhythmia, with the heart rate peaked at 189 beats per minute. The DCG and ECG were performed regularly to monitor her condition. The results were evaluated by a professional medical team and the medication dosage was adjusted according to the effect of treatment. She also suffered from upper respiratory infections several times during this period, but all had satisfactory outcomes after the treatment.
Although the arrhythmia and infections were well controlled, her clinical course was complicated by growth failure. The Griffiths Development Scales-Chinese Edition (GDS-C) and the Family Environment Scale Chinese Version (FES-CV) were performed for her at 30 months of age to evaluate her global development. The results showed that the total quotient of GDS-C was 66.71, and the total score of FES-CV was 54. In general, the results indicated that her overall development lagged, and her listening, language, as well as performance abilities, were weak. While the special examination showed that her muscle tone is normal, and her muscle strength level is V. The rehabilitation physician recommended her to do home rehabilitation training and professional rehabilitation training continuously. Moreover, it was worth noting that the result of the Screening Tool for Autism in Toddlers and Young Children (STAT) was high risk. Later, she often snores because of adenoid hypertrophy (Figure 2E), which may also be the cause of her poor sleep. In addition, her last visit was at 34 months for an allergy caused by eating crab, which improved after treatment (Figure 3).
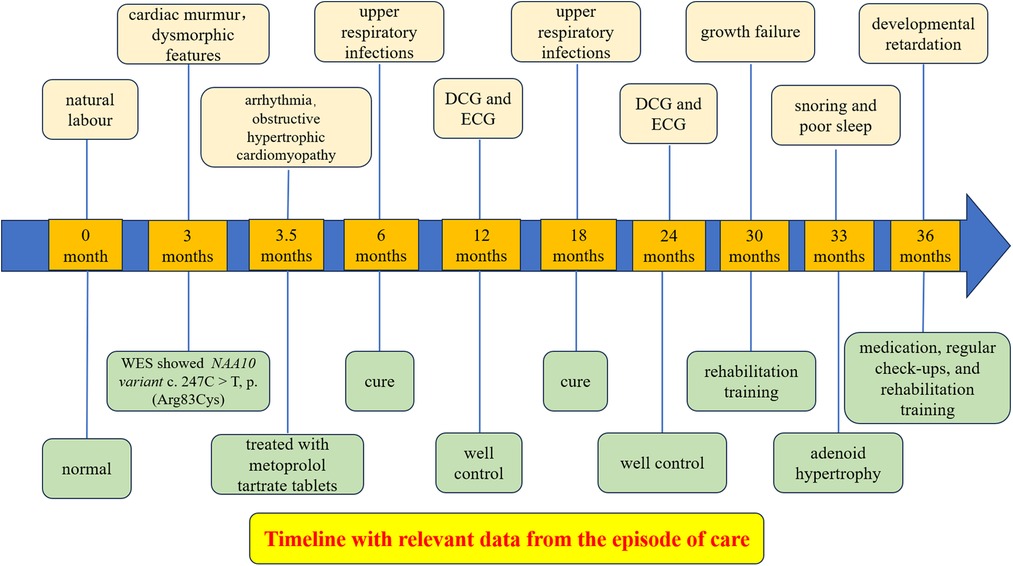
Figure 3. Timeline with relevant data from the episode of care.
Now she is 3 years old, and her overall development is about half a year behind normal. Recent cardiac color ultrasonography also suggests obstructive hypertrophic cardiomyopathy with moderate mitral regurgitation and mild tricuspid regurgitation. Although her global condition is well yet, medication, regular check-ups, and rehabilitation training under the guidance of a professional medical team are still required for her. In addition, the risk of autism also needs further evaluation or intervention.
Discussion
With the various NAA10 variants that have been identified, the phenotypic spectrum of individuals has vastly expanded in recent years. To date, 106 individuals with NAA10-related syndrome carrying 34 different de novo NAA10 missense variants have been reported (14). The NAA10-related syndrome is characterized by a broad spectrum of phenotypes in both males and females, ranging from asymptomatic girls to early lethality caused by structural or conductional cardiac abnormalities with the variable variant. The neurodevelopmental defects, including diverse degrees of ID, motor and language disability, autism spectrum disorder (ASD), and behavioral abnormities, were described as the most common features in all patients (15–17). In addition, facial dysmorphism is also universal but with no clear pattern. Moreover, postnatal growth retardation, impaired motor function, hypotonia, abnormal brain imaging, and recurrent infections were described frequently (9, 18). And in particular, cardiovascular defects were common, often leading to severe outcomes and in some cases fatal. However, due to the extremely limited number of clinical cases with each NAA10 variant, the genotype–phenotype relationship does not appear to be clear yet.
Furthermore, the levels of remaining enzymatic activity and the diverse function of NAA10 were discussed as the foundation underlying the NAA10-associated phenotypes (19–21). Nevertheless, a solid conclusion could not be drawn yet due to the less affected individuals. Functional research of diverse variants has been performed in previous studies, and the NatA complex stability as well as the global NatA acetyltransferase activity have been tested. Studies have shown that p. Val111Gly and p. Arg116Trp variants usually lead to mild phenotypes (6, 15). It is worth noting that different experimental approaches have been used in the functional characterization of NAA10 variants. For instance, Saunier et al. tested the enzymatic activity of purified monomeric NAA10 (not as part of the NatA complex) when studying the impact on Arg83Cys and Arg116Trp (6). When compared to the nearly abolished catalytic activity associated with the p. Phe128Leu, p. Phe128Ile, and p. Val107Phe variants, the partial catalytic impairment (about 60%) of the NAA10 subunit caused by the p. Arg83Cys variant was not reflected in different degrees of clinical phenotypes, suggesting the possibility of a critical threshold of enzyme activity necessary for normal function (6). Interestingly, in subsequent functional studies, Cheng et al. revealed that the NAA10 p. Arg83Cys variant leads to promoted NatA activity, suggesting that the phenotype of individuals with such variant might express via a particular mechanism (5).
In fact, the p. Arg83Cys variant identified in our patient is the most common variant and has been previously reported as pathogenic and occurs in a region conserved across species (10, 14). The phenotype described in this subtype usually consisted of variable degrees of neurodevelopmental defects, such as moderate to severe ID, motor and language disability, and ASD (5, 6). Feeding difficulties and postnatal failure to thrive with final short stature were described in approximately two-thirds of the patients. Facial dysmorphisms, including a prominent forehead, low-set ears, broad and flat nasal bridge, and particularly coarse face were depicted in more than half of the sufferings, while a specific pattern has not been identified. Furthermore, visual impairment, brain imaging anomalies, hypotonia, visual impairment, and recurrent infections were common too. What calls for special attention is that half of the patients exhibited different degrees of congenital structural cardiac abnormalities or arrhythmia (22). While, skeletal defects, hearing impairment, and seizures were described less frequently (22).
Overall, our patient confirmed the main clinical manifestations described previously such as dysmorphic features, developmental disorders, as well as congenital structural and conditional cardiac abnormalities. What's more, our patient also showed some rare but compatible features, including exophthalmos, blue sclera, and cutaneous capillary malformations. She also suffered from poor sleep, snoring, and facial changes caused by adenoid hypertrophy. The new rare clinical features expand the phenotype of NAA10-related syndrome. Her clinical course may enrich the knowledge of the trajectories and prognoses in patients with such conditions. Furthermore, given the fatal cardiac malformations or arrhythmias observed in the previous individuals, we recommend that patients should undergo precise medical follow-up, particularly for cardiac diseases (including hypertrophic cardiomyopathy and arrhythmias). In addition, her autistic tendencies also deserve further research to confirm.
It is well depicted that heterozygous females with NAA10-related syndrome have a wide spectrum of clinical manifestations, ranging from asymptomatic to varying degrees of developmental disorders and cardiac defects (6, 13, 22). It depends not only on the activity and stability of NatA acetyltransferase caused by specific variant types but also on X-chromosome skewing. Because most carrier mothers of boys affected with Ogden syndrome were asymptomatic, the X-chromosome inactivation (XCI) was almost completely skewed toward the wild-type allele (21). Given the present inability to speculate the genotype–phenotype correlations based on XCI, sufficient investigations should devote to exploring the effects of XCI on this syndrome to calculate its impact on phenotypic expression.
Conclusions
In summary, our patient confirmed the most common variant c. 247C > T, p. (Arg83Cys) and the main clinical manifestations such as dysmorphic features, developmental disorders, as well as congenital structural and conductional cardiac abnormalities. She also showed some rare but compatible features, including exophthalmos, blue sclera, adenoid hypertrophy, and cutaneous capillary malformations, which expand the phenotype of NAA10-related syndrome.
Data availability statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
KW wrote the first draft of the paper. CZ revised the paper and approved the final version for publication. All authors contributed to the article and approved the submitted version.
Funding
This project is supported by the National Natural Science Foundation of China (81670786) and Key R & D Projects of Zhejiang Provincial Science and Technology Agency (2021C03094).
Acknowledgments
We want to thank the patient's parents for their cooperation in providing the medical data and photographs necessary for this research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bienvenut WV, Sumpton D, Martinez A, Lilla S, Espagne C, Meinnel T, et al. Comparative large scale characterization of plant versus mammal proteins reveals similar and idiosyncratic N-alpha-acetylation features. Mol Cell Proteomics. (2012) 11(6):M111.015131. doi: 10.1074/mcp.M111.015131
2. Arnesen T, Van Damme P, Polevoda B, Helsens K, Evjenth R, Colaert N, et al. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc Natl Acad Sci U S A. (2009) 106(20):8157–62. doi: 10.1073/pnas.0901931106
3. Arnesen T, Anderson D, Baldersheim C, Lanotte M, Varhaug JE, Lillehaug JR. Identification and characterization of the human ARD1-NATH protein acetyltransferase complex. Biochem J. (2005) 386:433–43. doi: 10.1042/BJ20041071
4. Arnesen T, Starheim KK, Van Damme P, Evjenth R, Dinh H, Betts MJ, et al. The chaperone-like protein HYPK acts together with NatA in cotranslational N-terminal acetylation and prevention of huntingtin aggregation. Mol Cell Biol. (2010) 30(8):1898–909. doi: 10.1128/MCB.01199-09
5. Cheng H, Gottlieb L, Marchi E, Kleyner R, Bhardwaj P, Rope AF, et al. Phenotypic and biochemical analysis of an international cohort of individuals with variants in NAA10 and NAA15. Hum Mol Genet. (2020) 29(5):877–8. doi: 10.1093/hmg/ddz173
6. Saunier C, Stove SI, Popp B, Gerard B, Blenski M, AhMew N, et al. Expanding the phenotype associated with NAA10-related N-terminal acetylation deficiency. Hum Mutat. (2016) 37(8):755–64. doi: 10.1002/humu.23001
7. McTiernan N, Tranebjaerg L, Bjorheim AS, Hogue JS, Wilson WG, Schmidt B, et al. Biochemical analysis of novel NAA10 variants suggests distinct pathogenic mechanisms involving impaired protein N-terminal acetylation. Hum Genet. (2022) 141(8):1355–69. doi: 10.1007/s00439-021-02427-4
8. Rope AF, Wang K, Evjenth R, Xing JC, Johnston JJ, Swensen JJ, et al. Using VAAST to identify an X-linked disorder resulting in lethality in male infants due to N-terminal acetyltransferase deficiency. Am J Hum Genet. (2011) 89(1):28–43. doi: 10.1016/j.ajhg.2011.05.017
9. Popp B, Stove SI, Endele S, Myklebust LM, Hoyer J, Sticht H, et al. De novo missense mutations in the NAA10 gene cause severe non-syndromic developmental delay in males and females. Eur J Hum Genet. (2015) 23(5):602–9. doi: 10.1038/ejhg.2014.150
10. Sidhu M, Brady L, Tarnopolsky M, Ronen GM. Clinical manifestations associated with the N-terminal-acetyltransferase NAA10 gene mutation in a girl: ogden syndrome. Pediatr Neurol. (2017) 76:82–5. doi: 10.1016/j.pediatrneurol.2017.07.010
11. Hofman J, Hutny M, Chwialkowska K, Korotko U, Loranc K, Kruk A, et al. Case report: rare among ultrarare-clinical odyssey of a new patient with Ogden syndrome. Front Genet. (2022) 13:979377. doi: 10.3389/fgene.2022.979377
12. Gogoll L, Steindl K, Joset P, Zweier M, Baumer A, Gerth-Kahlert C, et al. Confirmation of Ogden syndrome as an X-linked recessive fatal disorder due to a recurrent NAA10 variant and review of the literature. Am J Med Genet A. (2021) 185(8):2546–60. doi: 10.1002/ajmg.a.62351
13. Wu YY, Lyon GJ. NAA10-related Syndrome. Exp Mol Med. (2018) 50(7):1–10. doi: 10.1038/s12276-018-0098-x
14. Lyon GJ, Vedaie M, Beisheim T, Park A, Marchi E, Gottlieb L, et al. Expanding the phenotypic spectrum of NAA10-related neurodevelopmental syndrome and NAA15-related neurodevelopmental syndrome. Eur J Hum Genet. (2023). doi: doi: 10.1038/s41431-023-01368-y. [Epub ahead of print].37130971
15. McTiernan N, Stove SI, Aukrust I, Marli MT, Myklebust LM, Houge G, et al. NAA10 Dysfunction with normal NatA-complex activity in a girl with non-syndromic ID and a de novo NAA10 p.(V111G) variant—a case report. BMC Med Genet. (2018) 19(1):47. doi: 10.1186/s12881-018-0559-z
16. Bader I, McTiernan N, Darbakk C, Boltshauser E, Ree R, Ebner S, et al. Severe syndromic ID and skewed X-inactivation in a girl with NAA10 dysfunction and a novel heterozygous de novo NAA10 p.(His16Pro) variant-a case report. BMC Med Genet. (2020) 21(1):153. doi: 10.1186/s12881-020-01091-1
17. Thevenon J, Duffourd Y, Masurel-Paulet A, Lefebvre M, Feillet F, El Chehadeh-Djebbar S, et al. Diagnostic odyssey in severe neurodevelopmental disorders: toward clinical whole-exome sequencing as a first-line diagnostic test. Clin Genet. (2016) 89(6):700–7. doi: 10.1111/cge.12732
18. Casey JP, Stove SI, McGorrian C, Galvin J, Blenski M, Dunne A, et al. NAA10 Mutation causing a novel intellectual disability syndrome with long QT due to N- terminal acetyltransferase impairment. Sci Rep. (2015) 5:16022. doi: 10.1038/srep16022
19. Esmailpour T, Riazifar H, Liu LN, Donkervoort S, Huang VH, Madaan S, et al. A splice donor mutation in NAA10 results in the dysregulation of the retinoic acid signalling pathway and causes lenz microphthalmia syndrome. J Med Genet. (2014) 51(3):185–96. doi: 10.1136/jmedgenet-2013-101660
20. Dorfel MJ, Lyon GJ. The biological functions of Naa10-from amino-terminal acetylation to human disease. Gene. (2015) 567(2):103–31. doi: 10.1016/j.gene.2015.04.085
21. Myklebust LM, Van Damme P, Stove SI, Dorfel MJ, Abboud A, Kalvik TV, et al. Biochemical and cellular analysis of Ogden syndrome reveals downstream Nt-acetylation defects. Hum Mol Genet. (2015) 24(7):1956–76. doi: 10.1093/hmg/ddu611
Keywords: NAA10, NAA10-related syndrome, n-terminal acetylation, ogden syndrome, case report
Citation: Wei K and Zou C (2023) Clinical manifestations in a Chinese girl with heterozygous de novo NAA10 variant c. 247C > T, p. (Arg83Cys): a case report. Front. Pediatr. 11:1198906. doi: 10.3389/fped.2023.1198906
Received: 2 April 2023; Accepted: 13 June 2023;
Published: 27 June 2023.
Edited by:
Paul Lasko, McGill University, CanadaReviewed by:
Miguel Angel Alcántara-Ortigoza, National Institute of Pediatrics, MexicoNina McTiernan, University of Bergen, Norway
© 2023 Wei and Zou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chaochun Zou emNjMTRAemp1LmVkdS5jbg==