 Bo Tan1,†
Bo Tan1,† Sihan Liu
Sihan Liu Xiaoshu Feng
Xiaoshu Feng Xin Pan
Xin Pan Xiaojing Dong
Xiaojing Dong- 1Department of Gynecology and Obstetrics, The Second Affiliated Hospital of Chongqing Medical University, Chongqing, China
- 2Institute of Rare Diseases, West China Hospital of Sichuan University, Chengdu, China
CCCTC-Binding Factor (CTCF) is a protein-coding gene involved in transcriptional regulation, insulator activity, and regulation of chromatin structure, and is closely associated with intellectual developmental disorders. In this study, we report two unrelated Chinese patients with intellectual disability (ID). According to variant interpretation results from exome sequencing data and RNA-seq data, we present two novel heterozygous CTCF variants, NM_006565.3:c.1519_2184del (p. Glu507_Arg727delins47) and NM_006565.3:c.1838_1852del (p.Glu613_Pro617del), found in two distinct unrelated patients, respectively. Moreover, RNA-seq data of patient 1 indicated the absence of the mutant transcript, while in patient 2, the RNA-seq data revealed a CTCF mRNA transcript with a deletion of 15 nucleotides. Notably, the RNA sequencing data revealed 507 differentially expressed genes shared between these two patients. Specifically, among them, 194 were down-regulated, and 313 were up-regulated, primarily involved in gene regulation and cellular response. Our study expands the genetic and clinical spectrum of CTCF and advances our understanding of the pathogenesis of CTCF in vivo.
Introduction
Intellectual disability (ID) is a neurodevelopmental disorder characterized by deficits in intelligence and adaptive functioning of varying severity before the age of 18. The global prevalence of ID is around 1%–3% in the general population and approximately 1% in China. ID can manifest as either an independent feature known as non-syndromic ID (NSID), or it can be accompanied by facial dysmorphic features and other morphological anomalies. Indeed, the majority of ID cases are attributed to genetic abnormalities. With the development of next-generation sequencing, researchers have identified more than 1,000 genes causally linked to ID (1–3).
CTCF (CCCTC-Binding Factor) is a protein-coding gene (OMIM: 604167) involved in a variety of chromatin regulatory processes, including gene expression, chromatin higher-order organization, and maintenance of chromatin 3D structure (4, 5). The CTCF gene consists of 12 exons that encode eleven zinc finger (ZF) domains. Numerous studies have identified to date 59 pathogenic variants in the CTCF gene associated with autosomal dominant ID, including 5 large deletion variants (1, 6–13). It is worth noting that most of these variants are de novo, meaning that they are newly occurring in the affected individuals and result in the loss-of-function of one copy of the CTCF gene. The association of CTCF with ID was first reported in 2013 by Gregor et al., who identified two de novo frameshift pathogenic variants, c.375dupT (p.Val126Cysfs14) and c.1186dupA (p.Arg396Lysfs13), as well as one de novo missense pathogenic variant, c.1699C>T (p.Arg567Trp), in CTCF in four syndromic ID patients (1). Subsequently, several studies reported numerous pathogenic CTCF variants in patients with ID and neurodevelopmental disorders (12, 14). While only three de novo CTCF disease-causing variants (c.615_618delGAAA[p.Lys206Profs15], c.1699C>T[p.Arg567Trp], c.329dupT[p.Gly111fs29) have been identified in Chinese patients associated with the neurodevelopmental disorder, there is an urgent need to report additional genetic and clinical features from Chinese ID patients to expand the spectrum of CTCF variation in the Chinese population (11).
Here, we present the genetic and clinical characterization of two unrelated Chinese ID patients with novel CTCF variants (NM_006565.3:c.1519_2184del (p. Glu507_Arg727delins47) and NM_006565.3:c.1838_1852del(p.Glu613_Pro617del). Additionally, we assess the disease-causing mechanism in our patients through RNA sequencing. This study expands current knowledge of the genetic and clinical spectrum of CTCF variants and reveals consistent phenotypes and disease mechanisms across populations.
Materials and methods
Subjects and clinical information
Patients were recruited from the Second Affiliated Hospital of Chongqing Medical University in Chongqing, China. The patients were clinically assessed, and medical experts and geneticists confirmed no other linked anomalies. This study was approved by the ethical committee of the Second Affiliated Hospital of Chongqing Medical University. Written informed consent was obtained from the patients' parents (approval number: 2023/458).
Exome sequencing
Genomic DNA from the patients was extracted from peripheral blood collected in EDTA tubes. Exome sequencing was performed on the patients using the Agilent (Santa Clara, CA) version 6 enrichment kit and the Illumina HiSeq 4000 sequencing system (paired-end reads, 2 × 150 bp).
Variants identification and interpretation
Trimmomatic was used to remove adapter contamination and trim low-quality reads to obtain clean reads (15). Then, the cleaned reads were aligned to the human reference genome (hg19) using the Burrows–Wheeler Alignment tool (16). DNA variants were called following the Genome Analysis Toolkit software best practices (17). Then, variants were annotated using Variant Effect Predictor (VEP) (18–22). Multiple computational predictive tools were applied to predict the pathogenicity of the detected variants (23–29). Further, variants with an allele frequency (AF) of <0.1% were retained for downstream analysis. According to the ACMG/AMP guidelines, all retained variants were classified into pathogenic (P), likely pathogenic (LP), variants with unknown clinical significance (VUS), likely benign (LB), or benign (B) (30). The putative diagnostic variants were experimentally validated by Sanger sequencing (ABI 3730xl Genetic Analyzer) and real-time PCR.
Copy number variants detection and interpretation
Copy number variations (CNV) were called using ExomeDepth (31) with default parameters, utilizing the BAM files generated from earlier. A set of 10 samples from our in-house control data served as the reference set. When examining potential CNV calls on the X chromosome, only samples of the same sex were considered for correlation. CNVs with a Bayes factor (BF) value below 50 were filtered out. Subsequently, each CNV call underwent additional annotation using the VEP, and classified according to the guidelines proposed by ACMG and ClinGen for CNV interpretation (32).
To validate the presence of CNVs, we performed CNV-seq analysis following the protocol proposed by Dong Zirui et al. (33). Briefly, mapped reads were grouped into 5 kb bins based on their mapped positions (hg19). The coverage of each bin was calculated using the mapped read depth within the bin and underwent a two-step bias correction (GC correction and population-level normalization). A CNV was considered a deletion if its average copy ratio was less than 0.6 or a duplication if it was greater than 1.4. Additionally, quantitative PCR (qPCR) was employed as an additional validation technique. The qPCR reactions were conducted using the SYBR Green I PCR Master Mix (Applied Biosystems) on the Applied Biosystems 7500 Fast Instrument.
RNA sequencing
Total RNA was extracted from the peripheral blood of these two patients and enriched by oligo-dT bead capture, separately. cDNA was synthesized according to the manufacturer's protocol, and cDNA libraries were constructed using the Illumina TruSeq stranded mRNA sample prep kit protocol (Illumina). Pooled samples were sequenced using a NovaSeq 6000 sequencing system.
Differential expression analysis
The clean RNA-sequencing reads were mapped to the human reference genome (hg 38) using STAR (2.7.8a) with the Gencode v29 annotation (34). Blood samples (n = 735) from GTEx v8 data were used as controls (35). Combat was applied to remove the batch effect between our data and GTEx data. Raw read counts were log-transformed by R package VOOM (36) first, filtering those with log2(CPM) < 0 in more than 75% of the samples. Differential expression analysis of the remaining genes was performed using the Limma (37) package while controlling for biological covariates and hidden factors identified by SVA (38). Gene was defined as a differentially expressed gene (DEG) with an adjusted p-value <0.05 and |log-transformed fold-change|>1. Pathway enrichment analysis of DEGs was carried out using KOBAS-i (39). The adjusted p-value cutoff for significant pathways was set at less than 0.05.
Validation of alternative splicing isoform
The RNA specimens collected from both the patients and the negative control underwent reverse transcription and subsequent amplification of cDNA using a forward primer specific to CTCF Exon 10 (5′-CTGCGGCTTTTGTCTGTTCT-3′) and a reverse primer specific to Exon 13 (5′-CCTCCTCTTCCTCTCCCTCT-3′). The PCR reaction was performed under the following conditions: an initial denaturation at 95°C for 3 min, followed by 40 cycles of denaturation at 95°C for 30 s, annealing at 58°C for 15 s, and extension at 72°C for 45 s. The resulting PCR products were then subjected to Sanger sequencing for further analysis.
Reverse transcription-quantitative PCR (Rt-qPCR)
Reverse transcription-quantitative PCR (RT-qPCR) was used to measure the relative expression of candidate differentially expressed genes in both patient and control blood cells. The GAPDH mRNA was utilized as an internal control, and the experiments were conducted in triplicate. Quantitative PCR (qPCR) analysis was performed using the QuantstudioTM 7 Flex system (Applied Biosystems) with the following amplification program: 95°C for 10 min, 40 cycles at 95°C for 15 s, and 60°C for 1 min.
Results
Genetic analysis of patient 1
Patient 1 is a 22-year-old female with a height of 146 cm (<3rd percentile) and a weight of 62 kg (89th percentile). She presented with moderate intellectual disability, strabismus, and scoliosis (Figure 1A). Exome sequencing identified a heterozygous 9,503 bp deletion in the genomic region of chr16:67,662,273–67,671,775 (hg19), specifically corresponding to NM_006565.3:c.1519_2184del (p.Glu507_Arg727delins47), in patient 1 (Figures 1C,D). This variant corresponds to a deletion spanning from exon 9 to exon 12 of the CTCF gene. As exome sequencing (ES) targets only the coding regions, to assess whether the intronic region of the CTCF gene was affected or not, we employed CNV-seq. The CNV-seq data indicated a 16,519 bp deletion in the region of chr16:67,658,826–67,675,344 (hg19), encompassing exon 8 to exon 12 of the CTCF gene (Supplementary Figure S1). Confirmation of the presence of the deletion of exon 9 to exon 12 in patient 1 was achieved through qPCR analysis, as illustrated in Figure 2A. Importantly, exon 8 of the CTCF gene and the adjacent regions downstream of the CTCF gene in patient 1 remained unaffected. This result revealed that the predicted deletion region by CNV-seq is attributed to the deletion of exons 9 to exon 12 and led us to speculate that the breakpoint for the c.1519_2184del variant may reside within the intronic or intergenic regions of the CTCF gene. However, the exact breakpoints of the deletion could not be identified. Furthermore, to investigate whether the CTCF mRNA in patient 1 underwent nonsense-mediated mRNA decay (NMD), we compared total CTCF RNA expression levels to controls. As shown in Figure 2B, the CTCF expression levels in patient 1 were 50% compared to controls, indicating that the mutant mRNA is undergoing NMD.
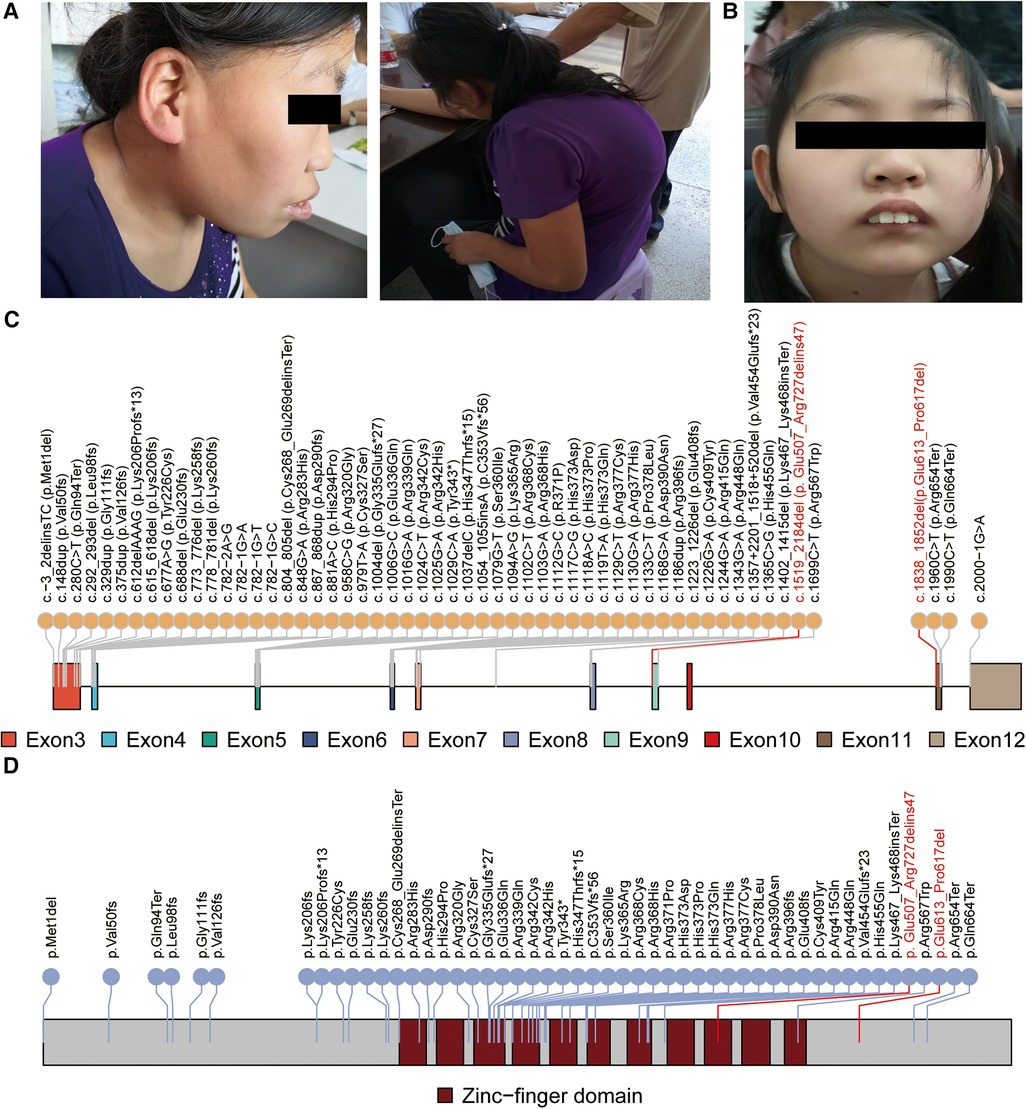
Figure 1. Genetic and clinical features of the patients. The clinical features of patient 1 (A) and patient 2 (B) are presented. Patient 1 is a 22-year-old female with moderate intellectual disability, strabismus, and scoliosis. Patient 2 is an 11-year-old female with mild intellectual disability, epilepsy, and distinct facial dysmorphic features, including prominent incisors and ptosis. Schematic figures (C,D) highlight the positions of the reported pathogenic variants and variants identified in this study (marked in red) within the CTCF mRNA and protein domain, respectively.
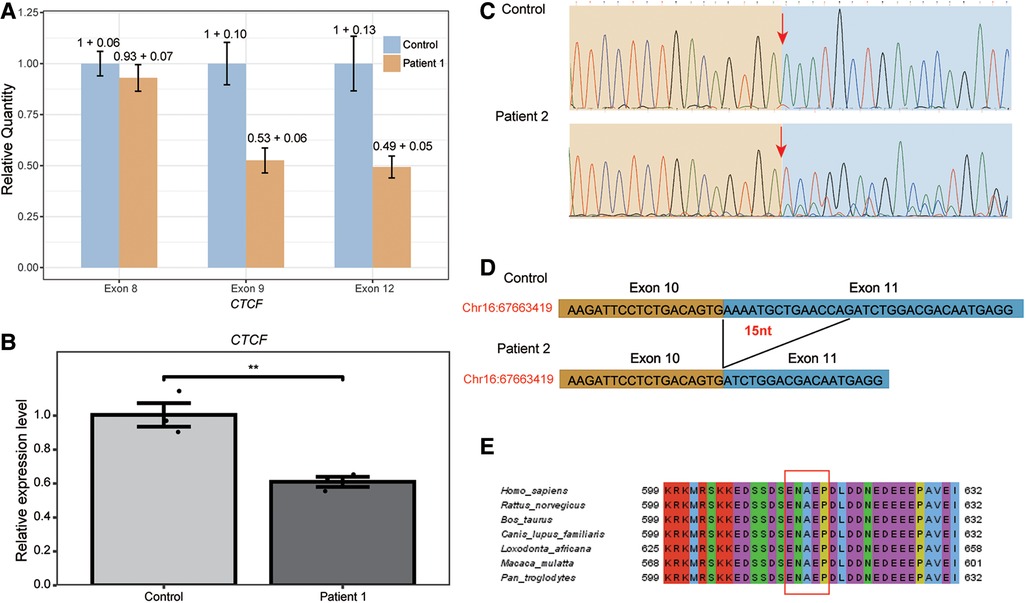
Figure 2. Validation of the two novel heterozygous CTCF deletion variants in patient 1 and in patient 2. (A) Real-time PCR was performed to validate the deletion of NM_006565.3:c.1519_2184del in the CTCF gene of patient 1. The relative quantity and standard deviation of exon 8, exon 9 and exon 12 in patient 1 and control samples was shown. (B) The expression of CTCF in patient 1 and control blood cells was measured using RT-qPCR, revealing that the CTCF expression levels in patient 1 were 50% compared to controls. This significant decrease indicates that the mutant mRNA is undergoing Nonsense-mediated mRNA decay (NMD). Statistical analysis showed a high level of significance relative to controls, with **p < 0.01. (C) The RNA specimens from both patient 2 and the negative control were subjected to reverse transcription and cDNA amplification. Sanger sequencing of the resulting PCR products was performed for detailed analysis. Our findings revealed that the CTCF c.1838_1852del variant led to a deletion of 15 nucleotides in the CTCF mRNA of patient 2. The arrow in the traces highlights the start of the deletion. (D) Schematic figures showing alternative splicing events caused by the c.1838_1852del. (E) The c.1838_1852del variant results in the loss of five amino acids (red box), which are located within a highly conserved region.
The CTCF gene is categorized as a haploinsufficient (HI) gene with a pLI score of 1. The deletion of exon 9 to exon 12 in the CTCF gene removes 30% (220/727) of the CTCF protein and could affect the zinc finger domain of the CTCF protein. Based on the ACMG/ClinGen guidelines for CNVs, the NM_006565.3:c.1519_2184del (p.Glu507_Arg727delins47) variant was classified as a likely pathogenic variant [criteria 2D-4 (0.9) = 0.9]. This classification was based on the variant's overlap with established HI genes and its result in exon deletions that include other exons in addition to the last exon, scoring 0.9 according to the guidelines. The inheritance status of this variant remains unknown as parental testing was not possible due to the family relocating, and her parents chose not to participate in the verification process.
Genetic analysis of patient 2
Patient 2 is an 11-year-old female weighing 32 kg (25th percentile), standing at a height of 135 cm (3rd percentile), with a head circumference of 52 cm (normal percentile). She exhibited mild intellectual disability, epilepsy, and distinctive facial dysmorphic features, including prominent incisors and ptosis. Notably, Patient 2 did not display any characteristic signs of skeletal dysplasia (Figure 1B). Exome sequencing identified a heterozygous splice site variant in the CTCF gene (NM_006565.3:c.1838-1_1841del) in patient 2. Sanger sequencing confirmed that the c.1838-1_1841del variant occurred de novo (Supplementary Figure S2), as it was not detected in the unaffected parents of patient 2 (PS2_supporting). In addition, this variant has not been annotated in several genomic databases (PM2), including dbSNP and gnomAD, and has not been reported in ClinVar.
To investigate whether the c.1838-1_1841del variant in patient 2 could affect mRNA splicing, we extracted and analyzed RNA from the patient's blood and a control sample using RT-PCR and RNA-sequencing. After separation on a 2% agarose gel, we observed a 516 bp band in both samples. Then, the RT–PCR products were purified and sequenced, and we observed that the c.1838-1_1841del variant caused a deletion of 15 nucleotides in the CTCF mRNA, leading to the deletion of 5 amino acids from the CTCF protein in patient 2 (Figures 2C,D). Furthermore, bioinformatics analysis revealed the deletion of five amino acid were located in the highly conserved region of exon 11 of the CTCF gene (Figure 2E). As an additional validation of the transcript result attributed to the c.1838-1_1841del variant, the RNA-seq data from patient 2 was visualized using the Gviz R package. Supplementary Figure S3 showcases the specific event that is associated with the c.1838-1_1841del variant. Based on these findings, it was appropriate to update the c.1838-1_1841del variant to c.1838_1852del (p.Glu613_Pro617del), accurately reflecting the 15-nucleotide deletion (Figures 1C,D). Considering the available evidence, the classification of the c.1838_1852del variant as a VUS is justified. Supporting criteria for this classification include PM4 (Protein length changes due to in-frame deletions/insertions), PM2, and PS2_supporting.
RNA-seq analysis
RNA-sequencing was conducted on RNA extracted from the blood cells of patient 1 and patient 2, both of whom had pathogenic CTCF variants, to elucidate the underlying mechanisms of the disease. A total of 14,254 expressed genes were included in the analysis, and 507 differentially expressed genes were identified with adjusted p-value <0.05 and |log-transformed fold-change|>1 threshold (Figure 3A and Supplementary Table S1). Among the 507 DEGs, 194 were down-regulated and 313 were up-regulated consistently and shared between the two patients, including several genes associated with neurodevelopmental disorders such as CAMKMT. Relative quantification of a subset of genes (CAMKMT, COX15, and GAS7) using RT-qPCR confirmed the accuracy of differential analysis with RNA-seq data (Figure 3B). Enrichment analyses of the DEGs revealed that they were involved in gene regulation and cellular response (Figure 3C and Supplementary Table S2).
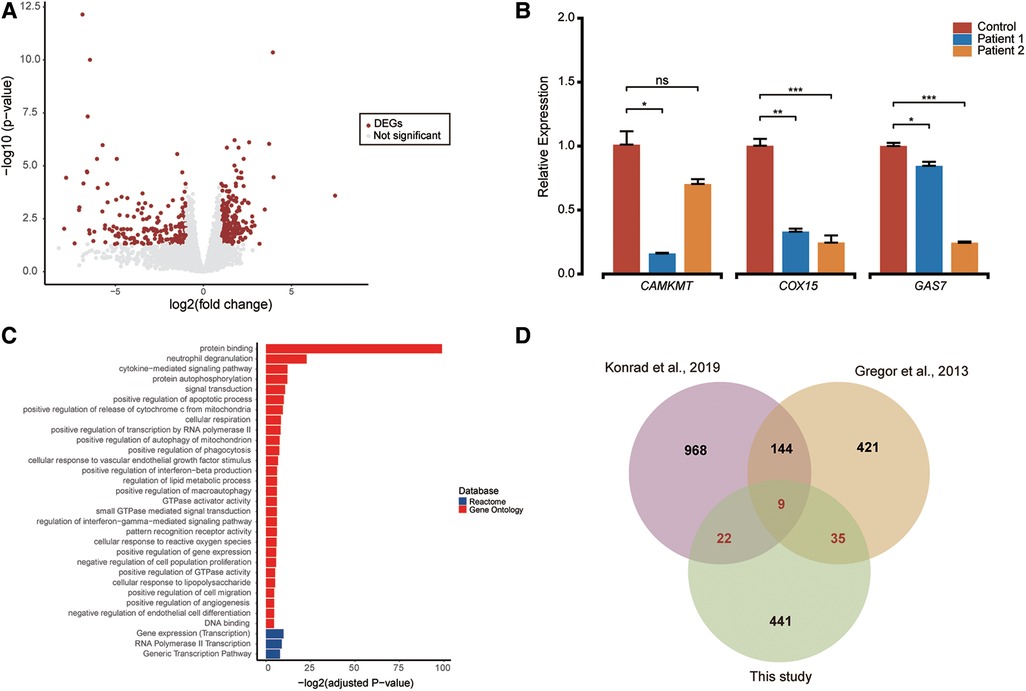
Figure 3. Differential expression analysis and functional enrichment (A). (A) The volcano plot showcases 507 differentially expressed genes (shown as red dots) out of 14,254 expressed genes, with an adjusted p-value < 0.05 and |log-transformed fold-change| > 1 threshold. (B) RT-qPCR quantification of candidate genes (CAMKMT, COX15, and GAS7) in patients and control blood cells. Two sample t-test was performed to test whether the expression of these genes between cases and control are significant difference or not. Relative normalized expression with standard deviation is presented on the Y-axis. Statistical significance is indicated as *p < 0.05, **p < 0.01, and ***p < 0.001. (C) Functional enrichment pathways of differentially expressed genes are shown with colors representing pathway items from various databases. The X-axis shows the log-transformed adjusted p value. Pathways with an adjusted p value <0.05 were selected as significant pathways and plotted. (D) The Venn plot illustrates the overlap of differentially expressed genes reported in this study compared to findings from two other papers. Differentially expressed genes were defined as genes with an adjusted p-value <0.05 and a |log-transformed fold-change| > 1.
Discussion
Identifying genetic causes of ID is crucial for understanding the molecular mechanisms underlying clinical features and for improving the management of the patients. In this study, we present two novel heterozygous CTCF variants NM_006565.3:c.1519_2184del (p. Glu507_Arg727delins47) and NM_006565.3:c.1838_1852del(p.Glu613_Pro617del) identified in two unrelated Chinese ID patients and validated the effect of these two variants on gene expression and splicing of the CTCF gene. However, it is important to note that the de novo status of the c.1519_2184del variant cannot be confirmed at this time due to the unavailability of parental testing for the patient. Our findings have not only identified the likely genetic causes of disease in these two patients but have also expanded the spectrum of disease-causing variants that can cause ID.
The CTCF gene is definitively associated with autosomal dominant syndromic ID, which was approved by the ClinGen Intellectual Disability and Autism Gene Curation Expert Panel on 7/21/2021 (40). A total of 54 pathogenic SNV/INDEL variants have been documented as causative for ID in the CTCF gene (Supplementary Table S3). These variants are primarily concentrated within exon 3 to exon 12 of the CTCF gene (Figure 1C) (1, 6–13). Furthermore, the majority of these variants are located in the zinc-finger domain and result in mild to moderate intellectual disability along with additional highly variable phenotypic features (Figure 1D). For instance, a female patient with a de novo c.1024C>T, p.Arg342Cys variant in the CTCF gene exhibited mild intellectual disability, a flat face, upslanting palpebral fissures, and a broad nose (13). Of note, previous studies have shown that the neurodevelopmental phenotype in individuals with larger deletions encompassing CTCF is not markedly different or more severe than in individuals with intragenic missense or likely gene-disruptive variants (12). This observation was consistent with our study, as the reported two patients showed mild or moderate ID. However, due to their rural background, many clinical investigations have not been conducted, and additional information on their early development, milestones, head circumference, cardiac anomalies, magnetic resonance imaging, and electroencephalography results is unavailable.
In our study, we report two novel heterozygous variants, NM_006565.3:c.1519_2184del (p. Glu507_Arg727delins47) and NM_006565.3:c.1838_1852del(p.Glu613_Pro617del), identified in unrelated Chinese patients with ID. The c.1519_2184del variant in patient 1 cause a partial deletion of the zinc-finger domain. We speculate that the breakpoint for the c.1519_2184del variant may reside within the intronic or intergenic regions of the CTCF gene since this variant affects exons 9–12 of the CTCF gene while leaving the nearby genes “CARMIL2” and “PARD6A” unaffected. Patient 2, carrying the c.1838_1852del variant, shares a notable clinical feature of prominent incisors with previously reported ID patients harboring pathogenic inframe variants in the CTCF gene (1, 7). We observed a difference in annotation based on the ES and RNA-seq data for the novel CTCF variant identified in patient 2. The discrepancy between the expected results based on the genomic annotation and the RNA-seq results in patient 2 might be attributed to alternative splicing or other post-transcriptional processes. The c.1838_1852del variant in the CTCF gene leads to a partial deletion of exon 11 in the CTCF mRNA. The deletion leads to the loss of 5 amino acids in a highly conserved region but does not impact the zinc-finger domain. Overall, the c. 1838_1852del variant in the CTCF gene may contribute to the patient 2's phenotype. Additional research and functional assays may be required to establish a more definitive clinical significance for the c. 1838_1852del variant.
RNA-seq provides an opportunity to investigate the molecular mechanisms underlying a patient's phenotype. In 2013, Gregor et al. used RNA sequencing to study three patients diagnosed with ID who carried pathogenic CTCF variants and discovered broad deregulation of genes involved in the cellular response to extracellular stimuli (1). Similarly, Konrad et al. identified 1,143 differentially expressed genes between five individuals with pathogenic CTCF variants and eight healthy controls (12). In our study, we also observed broad deregulation of genes (n = 507) in the patients. Among these, 66 DEGs were consistent when compared with the findings from the previous two studies. Moreover, nine genes (PELI1, CSF2RB, B3GNT5, SLC6A6, CPD, MEGF9, SOD2, CLEC7A, ABHD2) exhibited consistent differential expression across all three studies (Figure 3D). In addition, qPCR confirmed that many NDD-related genes, such as CAMKMT, COX15, and GAS7 involved in neurodevelopment and cancer, were downregulated in the patients (41–43). CAMKMT is linked to Hypotonia-Cystinuria Syndrome (OMIM #606407), which manifests as mild to moderate intellectual disability and respiratory chain complex IV deficiency. The deficiency of the COX15 gene leads to mitochondrial complex IV deficiency (OMIM #220110), presenting with encephalomyopathic features in the neonatal period for some patients, while others experience developmental regression within the first year of life. Additionally, GAS7 is associated with schizophrenia and plays a role in regulating neuronal migration and morphogenesis. Pathway enrichment analysis showed that the biological functions of these 507 DEGs were related to gene regulation and cellular response. Our study suggests that the main pathogenic mechanism of variants located in the CTCF domain is the imbalance of gene expression and regulation, which is consistent with previous research (1, 12).
In conclusion, our study demonstrates that autosomal dominant intellectual disability can be caused by exon deletion (c.1519_2184del) and inframe deletion variants (c.1838_1852del) in the CTCF gene. RT-PCR analysis confirmed that the c.1838_1852del variant led to the partial deletion of exon 11 from the CTCF mRNA, which may impact the function of the CTCF protein. Our study also adds to the existing knowledge of the genetic and clinical spectrum of CTCF variants in Chinese ID patients and highlights the consistent disease mechanisms of ID patients who carry pathogenic variants in the CTCF gene across different populations.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human: HRA005212). Publicly accessible at https://ngdc.cncb.ac.cn/gsa-human.
Ethics statement
The studies involving humans were approved by the Ethical Committee of the Second Affiliated Hospital of Chongqing Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
BT performed the experiments and wrote the manuscript. SL and XF performed RNA-seq analysis and wrote the manuscript. LL, GQ, XZ, and XP collected the clinical information of the patients and data analysis. HY and XD designed and supervised the study and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Scientific and Technological Research Program of Chongqing Municipal Education Commission (Grant No. KJQN202200420), “Kuanren talents” project of the Second Affiliated Hospital of Chongqing Medical University (13-003-003), Nan’an District Science and Health Joint Medical Scientific Research Project (2020-01), Program for Youth Innovation in Future Medicine, Chongqing Medical University (W0122), and Maternal and Child Health Research Cultivation Project of the Chongqing Health Commission (2023FY201).
Acknowledgments
We appreciate the patients and their family members for their participation in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2023.1195862/full#supplementary-material
References
1. Gregor A, Oti M, Kouwenhoven EN, Hoyer J, Sticht H, Ekici AB, et al. De novo mutations in the genome organizer CTCF cause intellectual disability. Am J Hum Genet. (2013) 93:124–31. doi: 10.1016/j.ajhg.2013.05.007
2. Vissers LE, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet. (2016) 17:9–18. doi: 10.1038/nrg3999
3. Ilyas M, Mir A, Efthymiou S, Houlden H. The genetics of intellectual disability: advancing technology and gene editing. F1000Res. (2020) 9. doi: 10.12688/f1000research.16315.1
4. Filippova GN, Fagerlie S, Klenova EM, Myers C, Dehner Y, Goodwin G, et al. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol Cell Biol. (1996) 16:2802–13. doi: 10.1128/MCB.16.6.2802
5. Rubio ED, Reiss DJ, Welcsh PL, Disteche CM, Filippova GN, Baliga NS, et al. CTCF physically links cohesin to chromatin. Proc Natl Acad Sci U S A. (2008) 105:8309–14. doi: 10.1073/pnas.0801273105
6. Iossifov I, O'roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. (2014) 515:216–21. doi: 10.1038/nature13908
7. Bastaki F, Nair P, Mohamed M, Malik EM, Helmi M, Al-Ali MT, et al. Identification of a novel CTCF mutation responsible for syndromic intellectual disability—a case report. BMC Med Genet. (2017) 18:68. doi: 10.1186/s12881-017-0429-0
8. Deciphering Developmental Disorders, S. Prevalence and architecture of de novo mutations in developmental disorders. Nature. (2017) 542:433–8. doi: 10.1038/nature21062
9. Hori I, Kawamura R, Nakabayashi K, Watanabe H, Higashimoto K, Tomikawa J, et al. CTCF deletion syndrome: clinical features and epigenetic delineation. J Med Genet. (2017) 54:836–42. doi: 10.1136/jmedgenet-2017-104854
10. Willsey AJ, Fernandez TV, Yu D, King RA, Dietrich A, Xing J, et al. De Novo coding variants are strongly associated with tourette disorder. Neuron. (2017) 94:486–99.e9. doi: 10.1016/j.neuron.2017.04.024
11. Chen F, Yuan H, Wu W, Chen S, Yang Q, Wang J, et al. Three additional de novo CTCF mutations in Chinese patients help to define an emerging neurodevelopmental disorder. Am J Med Genet C Semin Med Genet. (2019) 181:218–25. doi: 10.1002/ajmg.c.31698
12. Konrad EDH, Nardini N, Caliebe A, Nagel I, Young D, Horvath G, et al. CTCF variants in 39 individuals with a variable neurodevelopmental disorder broaden the mutational and clinical spectrum. Genet Med. (2019) 21:2723–33. doi: 10.1038/s41436-019-0585-z
13. Wang T, Kim CN, Bakken TE, Gillentine MA, Henning B, Mao Y, et al. Integrated gene analyses of de novo variants from 46,612 trios with autism and developmental disorders. Proc Natl Acad Sci U S A. (2022) 119:e2203491119. doi: 10.1073/pnas.2203491119
14. Barbosa M, Joshi RS, Garg P, Martin-Trujillo A, Patel N, Jadhav B, et al. Identification of rare de novo epigenetic variations in congenital disorders. Nat Commun. (2018) 9:2064. doi: 10.1038/s41467-018-04540-x
15. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. (2014) 30:2114–20. doi: 10.1093/bioinformatics/btu170
16. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
17. Mckenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010) 20:1297–303. doi: 10.1101/gr.107524.110
18. Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, et al. Human gene mutation database (HGMD): 2003 update. Hum Mutat. (2003) 21:577–81. doi: 10.1002/humu.10212
19. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. (2016) 536:285–91. doi: 10.1038/nature19057
20. Mclaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The ensembl variant effect predictor. Genome Biol. (2016) 17:122. doi: 10.1186/s13059-016-0974-4
21. Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. Clinvar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. (2018) 46:D1062–7. doi: 10.1093/nar/gkx1153
22. Liu X, Li C, Mou C, Dong Y, Tu Y. dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. (2020) 12:103. doi: 10.1186/s13073-020-00803-9
23. Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. (2003) 31:3812–4. doi: 10.1093/nar/gkg509
24. Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol. (2010) 6:e1001025. doi: 10.1371/journal.pcbi.1001025
25. Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. (2010) 20:110–21. doi: 10.1101/gr.097857.109
26. Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat. (2016) 37:235–41. doi: 10.1002/humu.22932
27. Jaganathan K, Kyriazopoulou Panagiotopoulou S, Mcrae JF, Darbandi SF, Knowles D, Li YI, et al. Predicting splicing from primary sequence with deep learning. Cell. (2019) 176:535–48.e24. doi: 10.1016/j.cell.2018.12.015
28. Yuan X, Bai J, Zhang J, Yang L, Duan J, Li Y, et al. CONDEL: detecting copy number variation and genotyping deletion zygosity from single tumor samples using sequence data. IEEE/ACM Trans Comput Biol Bioinform. (2020) 17:1141–53. doi: 10.1109/TCBB.2018.2876527
29. Steinhaus R, Proft S, Schuelke M, Cooper DN, Schwarz JM, Seelow D. Mutationtaster2021. Nucleic Acids Res. (2021) 49:W446–51. doi: 10.1093/nar/gkab266
30. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
31. Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. (2012) 28:2747–54. doi: 10.1093/bioinformatics/bts526
32. Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American college of medical genetics and genomics (ACMG) and the clinical genome resource (ClinGen). Genet Med. (2020) 22:245–57. doi: 10.1038/s41436-019-0686-8
33. Dong Z, Zhang J, Hu P, Chen H, Xu J, Tian Q, et al. Low-pass whole-genome sequencing in clinical cytogenetics: a validated approach. Genet Med. (2016) 18:940–8. doi: 10.1038/gim.2015.199
34. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. (2013) 29:15–21. doi: 10.1093/bioinformatics/bts635
35. Consortium, G.T. The GTEx consortium atlas of genetic regulatory effects across human tissues. Science. (2020) 369:1318–30. doi: 10.1126/science.aaz1776
36. Law CW, Chen Y, Shi W, Smyth GK. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. (2014) 15:R29. doi: 10.1186/gb-2014-15-2-r29
37. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. (2015) 43:e47. doi: 10.1093/nar/gkv007
38. Griffin AM, Cleveland HH, Schlomer GL, Vandenbergh DJ, Feinberg ME. Differential susceptibility: the genetic moderation of peer pressure on alcohol use. J Youth Adolesc. (2015) 44:1841–53. doi: 10.1007/s10964-015-0344-7
39. Bu D, Luo H, Huo P, Wang Z, Zhang S, He Z, et al. KOBAS-i: intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. (2021) 49:W317–25. doi: 10.1093/nar/gkab447
40. Riggs ER, Bingaman TI, Barry CA, Behlmann A, Bluske K, Bostwick B, et al. Clinical validity assessment of genes frequently tested on intellectual disability/autism sequencing panels. Genet Med. (2022) 24:1899–908. doi: 10.1016/j.gim.2022.05.001
41. Collins RL, Brand H, Redin CE, Hanscom C, Antolik C, Stone MR, et al. Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome. Genome Biol. (2017) 18:36. doi: 10.1186/s13059-017-1158-6
42. Meyer MA. Neuronal localization of GAS7 within human brain tissue: implications for schizophrenia research. Neurol Int. (2018) 10:7563. doi: 10.4081/ni.2018.7563
Keywords: RNA-seq, novel variant, CTCF, intellectual disability, clinical diagnosis
Citation: Tan B, Liu S, Feng X, Pan X, Qian G, Liu L, Zhang X, Yao H and Dong X (2023) Expanding the mutational and clinical spectrum of Chinese intellectual disability patients with two novel CTCF variants. Front. Pediatr. 11:1195862. doi: 10.3389/fped.2023.1195862
Received: 29 March 2023; Accepted: 7 August 2023;
Published: 17 August 2023.
Edited by:
Claudia Gonzaga-Jauregui, Universidad Nacional Autónoma de México, MexicoReviewed by:
Amanda Krause, University of the Witwatersrand, South AfricaAvinash Vijay Dharmadhikari, Children’s Hospital of Los Angeles, United States
© 2023 Tan, Liu, Feng, Pan, Qian, Liu, Zhang, Yao and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaojing Dong ZG9uZ3hpYW9qaW5nQGNxbXUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship