 Yuqing Xu
Yuqing Xu Linyan Zhu3
Linyan Zhu3 Yeqing Qian
Yeqing Qian Minyue Dong
Minyue Dong
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 19 June 2023
Sec. Genetics of Common and Rare Diseases
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1191068
This article is part of the Research Topic Rare Diseases Research and Diagnosis in Low- and Middle-Income Countries View all 36 articles
Introduction: Mutations of LAMA2 gene are associated with congenital muscular dystrophy (CMD). The LAMA2-related CMD mainly consists of two diseases, merosin deficient congenital muscular dystrophies type 1A (MDC1A) and limb girdle muscular dystrophy 23 (LGMD23). LGMD23 is characterized by slowly progressive proximal muscle weakness, which primarily affects the lower limbs and results in gait difficulties. Additional clinical features include increased serum creatine kinase, abnormal electromyography with or without white matter abnormalities on brain imaging.
Methods: Clinical data were collected from a Chinese Han family. Whole-exome sequencing, Sanger sequencing, RT-PCR and TA clone sequencing were performed on the family members.
Results: Compound heterozygous mutations of LAMA2: c.1693C > T (p. Q565*) (maternally inherited) and c.9212-6T > G (paternally inherited) were identified and confirmed in the proband. The mutation c.1693C > T (p. Q565*) was classified as pathogenic according to American College of Medical Genetics and Genomics (ACMG) guidelines. By performing RT-PCR and TA clone sequencing, an insertion of 40-bp intronic sequence (intron 64) was found in the transcripts of the proband and her father, which resulted in a frameshift and premature truncation codon of the LAMA2. In particular, the variant truncated the LamG domain of the LAMA2. Therefore, the c.9212-6T>G was classified as likely pathogenic according to American College of Medical Genetics and Genomics (ACMG) guidelines.
Discussion: Our findings described two novel mutations in a girl with LGMDR23, which contributes to the genetic counseling of the family and expands the clinical and molecular spectrums of the rare disease.
Laminin is a large family of heterotrimeric glycoproteins which contribute to cell adhesion, differentiation, neurite outgrowth and extracellular matrix architecture (1). Laminin subunit α2(LAMA2), belonging to the laminin family, interacts with integrin α7β1 (2, 3) and dystroglycan with the large globular(G) C-terminal domain. LAMA2 protein connects other laminins with its N-terminal domain to form a protein meshwork, which is helpful for the supramolecular assembly of the basal membrane (4, 5). The mutations in the LAMA2 gene caused congenital muscular dystrophy (CMD), an autosomal recessive disorder. The prevalence of CMDs are estimated to be 0.563 per 100,000 in Italy (6) and 0.017–0.083 per 100,000 in China (7). LAMA2-realted CMD ranges from severe, early onset merosin deficient congenital muscular dystrophies type 1A (MDC1A, OMIM 607855), to mild, limb-girdle muscular dystrophies (LGMDR23, OMIM 618138) (8, 9).
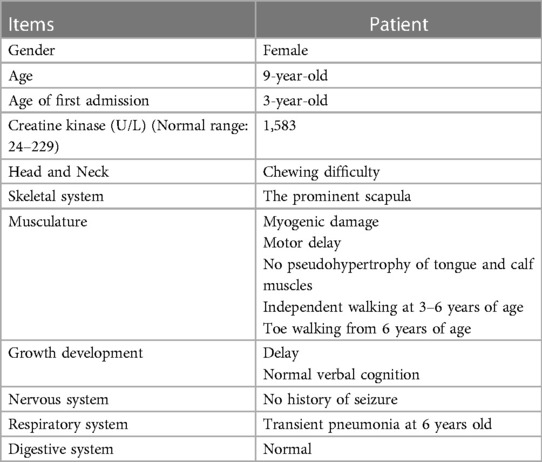
Table 1. Clinical and laboratory data of the proband.
Patients with MDC1A often suffer from hypotonia and severe proximal weakness at birth, as well as early respiratory and feeding difficulties. Furthermore, joint contractures, kyphoscoliosis and seizure are usually recognized. Compared with MDC1A, LGMDR23 is characterized by a milder and later age of onset, ranging from late childhood to mid-adulthood (10–12). Increased serum creatine kinase level, abnormal electromyography and abnormal white matter signal on brain magnetic resonance imaging (MRI) can be identified in both diseases.
Herein, we described a Chinese girl with LGMDR23 caused by two novel mutations in the LAMA2 gene.
A healthy non-consanguineous couple and their 9-year-old girl were referred to the Department of Reproductive Genetics, Women's Hospital, School of Medicine Zhejiang University in October, 2022. The girl (the proband) was born at term via virginal with an Apgar score of 10. Her development history was unremarkable until she was 3 years old. She was then firstly noted for being clumsy in general, as well as slow and awkward in her running. She had continued difficulties in jumping, running, lifting heavy objects and climbing stairs over time. Her serum creatine kinase was 1583 U/L (reference range: 24–229 U/L). Electromyography showed myogenic damage in limbs. Her language and learning development were normal. No family history of motor difficulties was observed. At the age of 6 years, she suffered from pneumoniae and recovered. Since then, she has achieved independent ambulation. In addition, she presents with toe walking and has difficulty in chewing due to masseter muscle weakness (Table 1).
This study was approved by the Institutional Review Board of the Women's Hospital, School of Medicine, Zhejiang University.
The whole-exome sequencing(WES) was performed as previously described (13).
The Sanger sequencing was conducted as previously described (13). The primers for c.1693C > T were 5′- ACAATGGAAGCCTATGTGAG -3′ and 5′- TGTGATTTAGCTGGTTCTGG -3′. The primers for c.9212-6T > G were 5′- ACACTTTGGGCATAGATGGG -3′ and 5′- CTTTGGTGAGCTTCAGGGAT -3′.
The RT-PCR experiment was conducted as previously described (13). The PCR primers were 5′-ACAACGACTGGAGTTCTTCT -3′ and 5′-TCCTGGGGTTACACTTATTT -3′. The PCR products were separated by electrophoresis on 2.0% agarose gel. Little difference was observed between the two mRNA splicing products, which caused difficulties for sequencing. TA cloning was chosen to purify the PCR products with the HieffCloneTM Zero TOPO-TA Cloning Kit (Yeasen, China). And then the purified PCR product was selected for sequencing.
The compound heterozygous variants of LAMA2: c.1693C > T (p.Q565*) and c.9212-6T > G were identified in the proband by whole-exome sequencing (Figure 1). Sanger sequencing confirmed that the c.1693C > T (p.Q565*) and c.9212-6T > G were inherited from the mother and the father, respectively. Therefore, the parents were both carriers and the co-segregation of genotype and phenotype was in accordance with autosomal recessive inheritance (PP4).
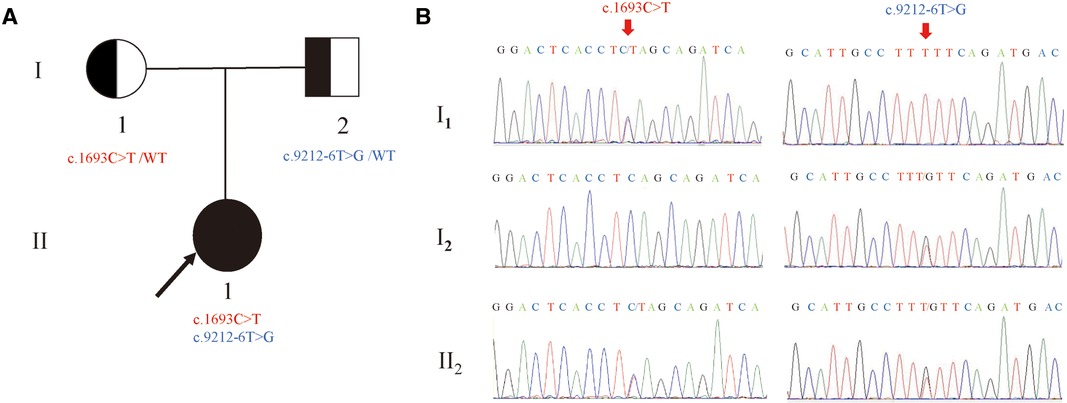
Figure 1. Newly identified nonsense and splicing variants in the LAMA2 gene. (A) The pedigree of the family.II1 (the proband) have two compound variants (c.1693C > T and c.9212-6T > G) in LAMA2 gene, which are inherited from the I1(mother) and the I2(father), respectively. (B) Sanger sequencing analysis. The two variants(c.1693C > T and c.9212-6T > G) were validated by Sanger sequencing. (red arrows indicated the mutation).
Neither of the variants had been reported in database (gnomAD, ClinVar or HGMD) or literature (PM2). The LAMA2: c.1693C > T (p.Q565*) variant theoretically introduces a stop codon and a truncated protein (PVS1). According to ACMG recommendations, the mutation LAMA2: c.1693C > T (p.Q565*) was classified as pathogenic (PP4 + PM2 + PVS1), while the variant LAMA2: c.9212-6T > G was classified as variant of uncertain significance (VUS).
NetGene2 Server (http://www.cbs.dtu.dk/services/ NetGene2/) and Alternative Splice Site Predictor (ASSP) (http://wangcomputing.com/assp/index.html) were used to predict the effect of the variant c.9212-6T > G on splicing. It was predicted that the variant affected splicing (Figure 2B–E). For the validation, RNAs were extracted from peripheral blood of the proband and her parents. cDNAs were then reverse transcribed to amplify exons 63–65 of LAMA2 with the primers. 2.0% agarose gel electrophoresis demonstrated that the proband and her father had a larger transcript than the normal amplification fragment (Figure 2A). Further results of TA clone sequencing and sequencing showed that the larger transcript had 40–bp intron 64 before the exon 65, which caused a truncation of LAMA2 by a frameshift and creation of a premature termination codon (Figure 2F). As was showed in Figure 2G, the splicing mutation (c.9212-6T > G) was predicted to generate prematurely truncated LAMA2 in the last LamG domain. The truncated effect does cause LAMA2 deficiency and damage the ability to interact with integrin α7β1 and dystroglycan. Considering the accordance between phenotype and genotype and the rarity of the splicing variant, the mutation LAMA2: c.9212-6T > G was classified as likely pathogenic (PP4 + PM2 + PS3) according to ACMG guidelines.
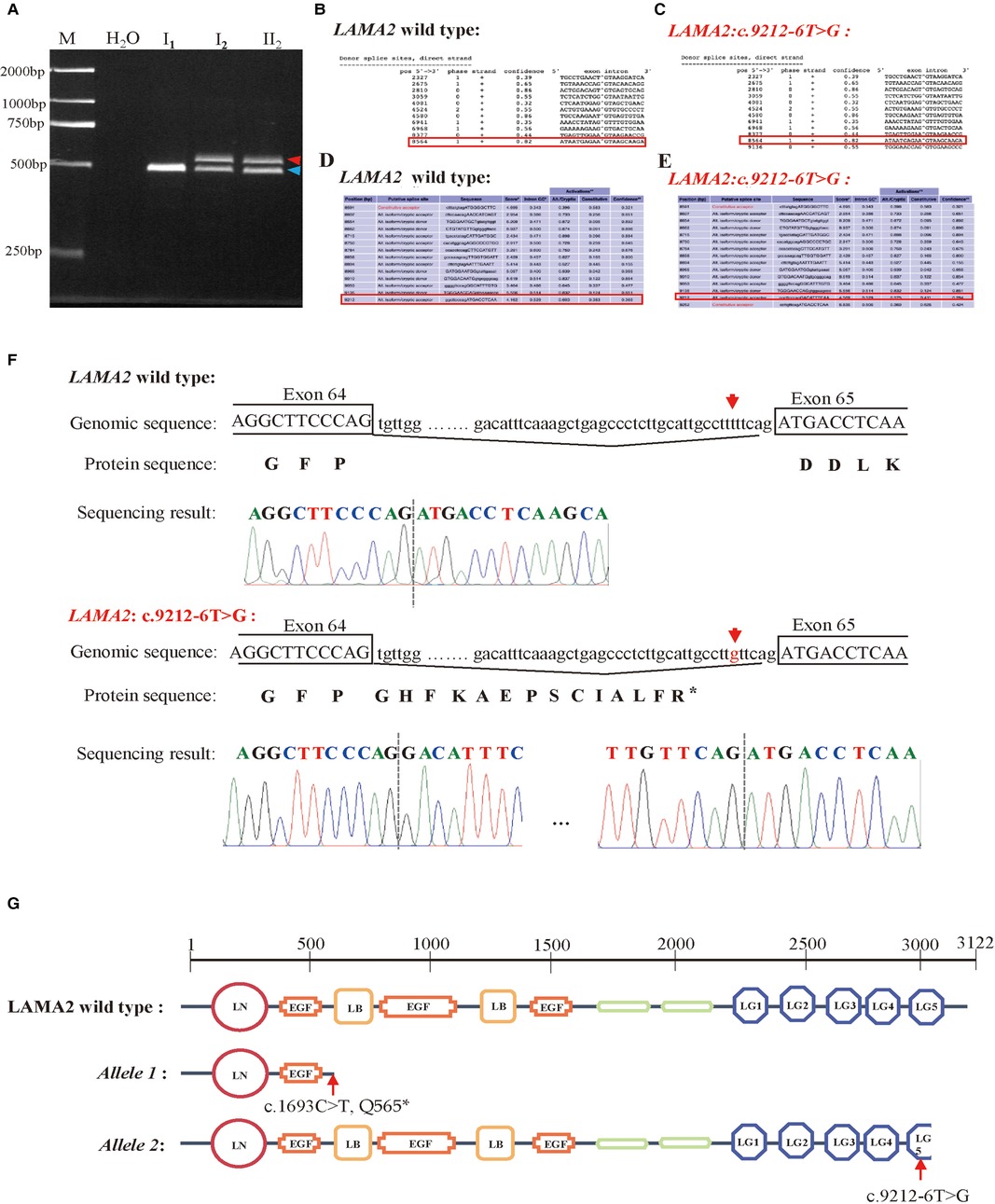
Figure 2. Analysis of the c.9212-6T > G variant in the LAMA2 gene. (A) RT-PCR analysis of LAMA2 cDNA from peripheral blood samples of the family members. Agarose gel (2.0%) electrophoresis of RT-PCR products generated from I1(mother), I2(father) and II1(the proband). Aberrantly spliced mRNA and normal spliced mRNA are marked by red and blue arrowheads, respectively. (B–E) Predictive results of the c.9212-6T > G variant site in splicing. (B) The Predictive result of wild type LAMA2 by using NetGene2 Server (the red square represented). (C) The Predictive result of LAMA2: c.9212-6T > G by using NetGene2 Server (the red square represented). (D) The Predictive result of wild type LAMA2 by using ASSP tool (the red square represented). (E) The Predictive result of LAMA2: c.9212-6T > G by using ASSP tool (the red square represented). (F) TA clone sequencing and sequencing analysis of the RT-PCR products from the proband. The red arrows reveal the position of the c.9212-6T > G variant. (G) Diagrams of domains of wild-type, c.1693C > T (p.Q565*) variant and c.9212-6T > G variant of LAMA2. The LAMA2: c.1693C > T (p. Q565*) has only two domains and the LG5 domain of LAMA2: c.9212-6T > G is truncated compared with wild type LAMA2.
In the current investigation, we reported a Chinese girl diagnosed as LGMDR23 due to the compound heterozygous variants in the LAMA2 gene. The maternally inherited mutation c.1693C > T (p.Q565*) of LAMA2 was pathogenic and the paternally inherited mutation c.9212-6T > G was proved to be a splicing mutation which resulted in frameshift and truncation. To the best of our knowledge, both of the variants have never been reported in any database or literature, indicating our findings extend the mutation spectrum and clinical knowledge of the diagnosis for the LGMDR23.
The patients with CMD due to laminin anomaly presented with severe muscle weakness, hypotonia and white matter abnormalities in brain imaging (14–16). In addition, all the patients could not achieve independent ambulation. More variabilities in the phenotype associated with LAMA2 deficiency were described thereafter. Compared with the severe features, some patients were characterized by milder and later-onset muscle weakness, slightly elevated creatine kinase level, ability to achieve ambulation, with or without abnormal brain imaging (17, 18). By carrying out muscle biopsies and immunocytochemistry with antibodies against 80 and 300 kDa fragment of LAMA2, complete absence of LAMA2 expression was found in patients with severe phenotype while reduced LAMA2 expression was found in those patients with mild phenotype. Therefore, LAMA2-related muscular dystrophy (MD) is generally divided into MDC1A (also named LAMA2-CMD, the severe one) and LGMDR23(the mild one) categories.
LGMDR23 is much rarer than MDC1A in China. Even in the largest and multicenter research of LAMA2-related muscular dystrophy in 2021, 14 cases of LGMDR23 were reported while 116 cases of MDC1A were reported (19). The symptoms of LGMDR23 included myopathic gait, difficulties in running and jumping, and epilepsy. Moreover, the median age of onset for LGMDR23 was 18.0 months (range 13.0–156.0 months) (19). In the current investigation, the proband was firstly noted for being clumsy and being slow and awkward in her running at 3 years old, belonging to the late-onset of LAMA2-related MD. She also had other typical symptoms like elevated creatine kinase level, transient pneumonia, delayed motor milestones, difficulty in chewing and abnormal electromyography. Additionally, the proband presented with toe walking which was also observed in a Turkish origin male at 10 years old (20) and in a Chinese origin female at 4 years old and a male at 6 years old (21).
The variants c.1693C > T (p.Q565*) and c.9212-6T > G of LAMA2 gene were detected by WES and confirmed by Sanger sequencing. These two variants were inherited from her mother and father, respectively. Based on ACMG guidelines, the nonsense c.1693C > T (p.Q565*) variant was classified as pathogenic while the c.9212-6T > G was classified as VUS. To date, approximately 100 nonsense variants and 90 splicing variants of LAMA2 have been described, without obvious mutational hotspots (22). After analysis of the genotype-phenotype correlations of the MDC1A and LGMDR23, Dandan et al. concluded that nonsense, frameshift or copy numbers variant caused more damage than splicing or missense variants (19).
With the highly genotype-phenotype correlation and predicted results of NetGene2 Server and ASSP, RT-PCR, TA clone sequencing and sequencing were performed to investigate the pathology of the splicing mutation. The c.9212-6T > G variant led to 40-bp intron insertion before the exon 65, which caused frameshift and truncation of the LAMA2 (Figure 2F). Thus, the significant LamG domain at the C-terminus of LAMA2 was truncated, which might damage the linkage between the extracellular matrix and the dystrophin-glycoprotein. The results upgraded the pathogenicity evidence of the c. c.9212-6T > G variant, so the variant classification was changed from VUS to likely pathogenic. In the meanwhile, because the splicing variant still allow partial expression of LAMA2 gene, it might explain the mild and late on-set phenotype in the proband like those patients in a previous published study (23). However, muscle biopsy was not performed for further study because we could not get the permission from the family to obtain the sample.
In summary, we described a Chinese girl diagnosed as LGMDR23 due to typical clinical phenotypes and laboratory tests, and identified two novel variants of LAMA2 gene via WES and Sanger sequencing. By performing RT-PCR,TA clone sequencing, we demonstrated the variant c.9212-6T>G was a likely pathogenic splicing mutation. Our findings expand the mutation spectrum and provided information for the genetic counseling of LGMDR23.
The original contributions presented in the study are publicly available. This data can be found here: https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA013814.
The studies involving human participants were reviewed and approved by Institutional Review Board of the Women's Hospital, School of Medicine, Zhejiang University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
MD conceived of the study, participated in its design and revised the manuscript; YX carried out the RT-PCR and drafted the manuscript; LZ analyzed the clinical data; YQ collected clinical data and revised the manuscript; All authors have read and approved the final manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (grant numbers 82171848), Zhejiang Provincial Natural Science Foundation of China (grant numbers LY22H110004) and the National Natural Science Foundation of China (grant numbers 81901382).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tzu J, Marinkovich MP. Bridging structure with function: structural, regulatory, and developmental role of laminins. Int J Biochem Cell Biol. (2008) 40(2):199–214. doi: 10.1016/j.biocel.2007.07.015
2. Vachon PH, Xu H, Liu L, Loechel F, Hayashi Y, Arahata K, et al. Integrins (alpha7beta1) in muscle function and survival. Disrupted expression in merosin-deficient congenital muscular dystrophy. J Clin Invest. (1997) 100(7):1870–81. doi: 10.1172/JCI119716
3. McKee KK, Harrison D, Capizzi S, Yurchenco PD. Role of laminin terminal globular domains in basement membrane assembly. J Biol Chem. (2007) 282(29):21437–47. doi: 10.1074/jbc.M702963200
4. Yurchenco PD, Schittny JC. Molecular architecture of basement membranes. FASEB J. (1990) 4(6):1577–90. doi: 10.1096/fasebj.4.6.2180767
5. Yurchenco PD, Cheng YS, Colognato H. Laminin forms an independent network in basement membranes. J Cell Biol. (1992) 117(5):1119–33. doi: 10.1083/jcb.117.5.1119
6. Graziano A, Bianco F, D’Amico A, Moroni I, Messina S, Bruno C, et al. Prevalence of congenital muscular dystrophy in Italy: a population study. Neurology. (2015) 84(9):904–11. doi: 10.1212/WNL.0000000000001303
7. Ge L, Zhang C, Wang Z, Chan SHS, Zhu W, Han C, et al. Congenital muscular dystrophies in China. Clin Genet. (2019) 96(3):207–15. doi: 10.1111/cge.13560
8. Oliveira J, Gruber A, Cardoso M, Taipa R, Fineza I, Gonçalves A, et al. LAMA2 Gene mutation update: toward a more comprehensive picture of the laminin-α2 variome and its related phenotypes. Hum Mutat. (2018) 39(10):1314–37. doi: 10.1002/humu.23599
9. Straub V, Murphy A, Udd B. 229th ENMC international workshop: limb girdle muscular dystrophies—nomenclature and reformed classification Naarden, the Netherlands, 17–19 March 2017. Neuromuscul Disord. (2018) 28(8):702–10. doi: 10.1016/j.nmd.2018.05.007
10. Hayashi YK, Ishihara T, Domen K, Hori H, Arahata K. A benign allelic form of laminin alpha 2 chain deficient muscular dystrophy. Lancet. (1997) 349(9059):1147. doi: 10.1016/S0140-6736(05)63023-1
11. Naom I, D’Alessandro M, Sewry CA, Philpot J, Manzur AY, Dubowitz V, et al. Laminin alpha 2-chain gene mutations in two siblings presenting with limb-girdle muscular dystrophy. Neuromuscul Disord. (1998) 8(7):495–501. doi: 10.1016/S0960-8966(98)00065-0
12. Rajakulendran S, Parton M, Holton JL, Hanna MG. Clinical and pathological heterogeneity in late-onset partial merosin deficiency. Muscle Nerve. (2011) 44(4):590–3. doi: 10.1002/mus.22196
13. Chen S, Xu Y, Qian Y, Li Z, Dong M. Case report: novel splicing mutations in RFX5 causing MHC class II deficiency. Front Genet. (2022) 13:978688. doi: 10.3389/fgene.2022.978688
14. Tomé FM, Evangelista T, Leclerc A, Sunada Y, Manole E, Estournet B, et al. Congenital muscular dystrophy with merosin deficiency. C R Acad Sci III. (1994) 317(4):351–7. PMID: 8000914
15. Helbling-Leclerc A, Zhang X, Topaloglu H, Cruaud C, Tesson F, Weissenbach J, et al. Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat Genet. (1995) 11(2):216–8. doi: 10.1038/ng1095-216
16. Dubowitz V, Fardeau M. Proceedings of the 27th ENMC sponsored workshop on congenital muscular dystrophy. 22–24 April 1994, The Netherlands. Neuromuscul Disord. (1995) 5(3):253–8. doi: 10.1016/0960-8966(95)90011-X
17. Herrmann R, Straub V, Meyer K, Kahn T, Wagner M, Voit T. Congenital muscular dystrophy with laminin alpha 2 chain deficiency: identification of a new intermediate phenotype and correlation of clinical findings to muscle immunohistochemistry. Eur J Pediatr. (1996) 155(11):968–76. doi: 10.1007/BF02282889
18. Naom IS, D’Alessandro M, Topaloglu H, Sewry C, Ferlini A, Helbling-Leclerc A, et al. Refinement of the laminin alpha2 chain locus to human chromosome 6q2 in severe and mild merosin deficient congenital muscular dystrophy. J Med Genet. (1997) 34(2):99–104. doi: 10.1136/jmg.34.2.99
19. Tan D, Ge L, Fan Y, Chang X, Wang S, Wei C, et al. Natural history and genetic study of LAMA2-related muscular dystrophy in a large Chinese cohort. Orphanet J Rare Dis. (2021) 16(1):319. doi: 10.1186/s13023-021-01950-x
20. Nelson I, Stojkovic T, Allamand V, Leturcq F, Bécane HM, Babuty D, et al. Laminin α2 deficiency-related muscular dystrophy mimicking emery-dreifuss and collagen VI related diseases. J Neuromuscul Dis. (2015) 2(3):229–40. doi: 10.3233/JND-150093
21. Nguyen NL, Ngoc CTB, Vu CD, Nguyen TTH, Nguyen HH. Whole exome sequencing as a diagnostic tool for unidentified muscular dystrophy in a Vietnamese family. Diagnostics (Basel). (2020) 10(10):741–10. doi: 10.3390/diagnostics10100741
22. Beytía Mde L, Dekomien G, Hoffjan S, Haug V, Anastasopoulos C, Kirschner J. High creatine kinase levels and white matter changes: clinical and genetic spectrum of congenital muscular dystrophies with laminin alpha-2 deficiency. Mol Cell Probes. (2014) 28(4):118–22. doi: 10.1016/j.mcp.2013.11.002
Keywords: LAMA2, LAMA2-related CMD, limb girdle muscular dystrophy 23, whole exome sequencing, LamG domain
Citation: Xu Y, Zhu L, Qian Y and Dong M (2023) Limb girdle muscular dystrophy 23 caused by compound heterozygous mutations of LAMA2 gene. Front. Pediatr. 11:1191068. doi: 10.3389/fped.2023.1191068
Received: 21 March 2023; Accepted: 5 June 2023;
Published: 19 June 2023.
Edited by:
Vajira Dissanayake, University of Colombo, Sri LankaReviewed by:
Musharraf Jelani, Islamia College University, Pakistan© 2023 Xu, Zhu, Qian and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Minyue Dong ZG9uZ215QHpqdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.