 Xuliang Zhao1
Xuliang Zhao1 Xu Li
Xu Li Weiwei Sun
Weiwei Sun Ruixia Tian
Ruixia Tian
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 19 May 2023
Sec. Genetics of Common and Rare Diseases
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1180381
Background: The p120-ctn protein, encoded by CTNND1, is involved in intercellular connections and regulates epithelial–mesenchymal transformation. CTNND1 mutations can lead to blepharocheilodontic syndrome (BCDS). Increasing evidence shows that although BCDS mainly manifests as craniofacial and oral deformities, it can also present as congenital heart disease, limb deformities, and neurodevelopmental disorders.
Case description: We report a prenatal case of a major cardiac malformation at 24+3 weeks of gestation. Ultrasound examination revealed a hypoplastic left ventricular, aortic coarctation, and a ventricular septal defect. Genetic analysis of the fetal tissues showed the presence of a novel mutation in CTNND1 (NM_001085458.2: c.566_c.567insG; p.Pro190fs*15), which may lead to premature termination of protein coding, while both the parents harbored wild-type CTNND1. To date, only 15 CTNND1 mutations have been reported in 19 patients worldwide, of which approximately 31% (6/19) had a cardiac phenotype.
Conclusion: To the best of our knowledge, this is the first case report of fetal complicated cardiac malformations caused by this CTNND1 mutation. Our findings provide new clinical references for prenatal diagnosis and suggest an important role for CTNND1 in early cardiac development.
Blepharocheilodontic syndrome (BCDS) is a rare autosomal dominant genetic disease, clinical manifestations of which consist of ectodermal dysplasia, such as eyelid deformity, cleft lip and palate, and abnormal teeth, as well as hypothyroidism, anal atresia, and neural tube defects caused by CTNND1 mutations (1, 2). There are two subtypes of BCDS: BCDS1 (OMIM #119580), caused by CDH1 mutations, and BCDS2 (OMIM #617681), caused by CTNND1 mutations. These two genes may be involved in E-cadherin degradation and lead to BCDS, resulting in two BCDS subtypes with relatively similar phenotypes, both being mainly characterized by craniofacial deformities (3). Compared to the BCDS phenotype in humans, that in animal models can play a role in more extensive developmental abnormalities. For example, abnormal development of teeth, blood vessels, the kidneys, and the pancreas was observed in the Ctnnd1 conditional knockout mouse model (4–6). Therefore, an increase in the number of reported patients with BCDS may enrich the knowledge of the disease phenotype. To date, less than 20 patients with CTNND1 mutations have been reported. Alharatani et al. reported 13 patients with CTNND1 mutations in nine families and expanded the range of phenotypes, with 6 patients presenting with congenital heart disease (CHD) of differing severities (7).
Here, we report a prenatal case of major fetal heart disease. The gene test results from fetal tissue after labor induction indicated a novel CTNND1 frameshift mutation. The findings from this prenatal case provide clinical resources for prenatal diagnosis and genetic counseling and enrich the reported disease phenotypes and mutation spectrum.
A 25-year-old Han Chinese pregnant woman (first pregnancy) underwent an ultrasound examination at 24+3 weeks of gestation. The ultrasound results of the fetal heart showed a hypoplastic but apex-forming left ventricle, a hypoplastic aortic arch, coarctation of the aorta, and a ventricular septal defect (Figure 1). After induction of labor, fetal autopsy revealed ptosis and no obvious limb abnormalities, and the cardiac diagnosis was confirmed, per the suspicion arising after the ultrasound analysis.
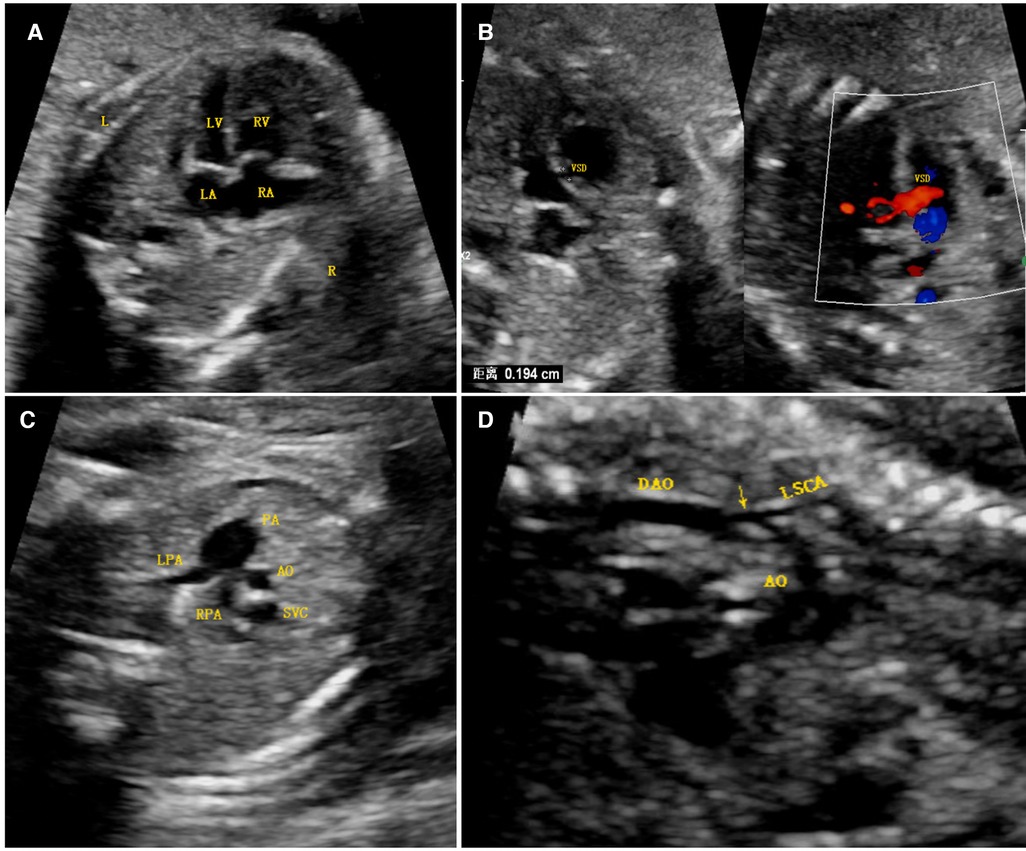
Figure 1. Antenatal ultrasound image. (A) The four-chamber view shows that the right ventricular cavity was significantly larger than the left cardiac cavity, the latter of which was still apex forming. (B) A ventricular septal defect was found just below the aortic outflow, as seen via two-dimensional imaging and color Doppler imaging. (C) A three-vessel view demonstrated a significantly larger main pulmonary artery relative to the aorta (PA > 1.6 AO), the latter of which was smaller than the superior vena cava. (D) A long-axis image of the aortic arch demonstrated diffuse hypoplasia of the arch to the isthmus and a posterior shelf (arrow), in keeping with the aortic coarctation. Abbreviations: LV, left ventricle; RV, right ventricle; LA, left atrium; VSD, ventricular septal defect; RA, right atrium; PA, pulmonary artery; LPA, left pulmonary artery; RPA, right pulmonary artery; SVC, superior vena cava; DAO, descending aorta; LSCA, left subclavian artery; AO, aorta.
Both parents were healthy and of Han Chinese ethnicity, were not close relatives, and denied a family history of congenital heart disease. They authorized the genetic testing of the fetal tissue to identify the pathogenic factors of this fetal CHD.
We obtained written informed consent from the parents for publication of the clinical data, image map, and genetic mutation data of the fetus. All procedures were performed in accordance with the Declaration of Helsinki of 1964 and reviewed by the Ethics Committee of the 901st Hospital of the Joint Service of the People's Liberation Army (batch number 202112001).
We collected approximately 0.5 cm × 1.0 cm of skin tissue from the inner side of the fetus left leg; genomic DNA was extracted from the tissue samples, fragmented to an average size of 180–280 bp, and then used to create a DNA library according to the established Illumina paired-end scheme. The IDT xGen Exome Research Panel v2 (Integrated DNA Technologies, Coralville, IA, USA) was used for whole-exome sequencing, according to the manufacturer's instructions. The Illumina Novaseq 6,000 platform (Illumina Inc., San Diego, CA, USA) was used to sequence the genomic DNA with a minimum coverage depth of 10× and a target coverage of 99% (average coverage depth of 110×).
Offline data uses bcl2fastq v.2.20.0 software (Illumina Inc.) for basic call file conversion and demultiplexing. The BWA algorithm was used to align all data with the reference sequence (hg19/GRCh37). ANNOVAR (https://annovar.openbioinformatics.org/en/latest/user-guide/download/) was used to annotate the secondary allele frequencies, and toxicity and protection scores from the public dataset was used to filter and evaluate the possible pathogenicity of the variation. Variation was filtered according to public databases (dbSNP, 1,000 Genomes, and gnomAD), and that reported as pathogenic or possibly pathogenic in the Human Gene Mutation Database (http://www.hgmd.org/) and ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) was retained. Online bioinformatics tools (SIFT, PolyPhen2, and MutationTaster) were used to predict the biohazards of the variation. Candidate pathogenic variants were identified according to the function, variation, and genetic pattern of the genes, and further evaluated according to the American College of Medical Genetics and Genomics guidelines (8).
The primers for the exome-coding region of the target gene were designed using the Primer Premier software (version 5.0; Premier Bio-soft International, Palo Alto, CA, USA). The primer sequences for CTNND1 were as follows: F, 5′-CCGGGAGAGACTGCCATAGTTA-3′; R, 5′-CTGTGACACGAGAGACAGTACA-3′. PCR was performed using PCR MasterMix polymerase (Tianjin Biotech, Beijing, China). PCR products were directly sequenced on an ABI 3,500 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
We identified a frameshift mutation in exon 6 of CTNND1, namely NM_001085458.2: c.566_c.567insG (p.Pro190fs*15), in the fetal tissue. Both parents carry wild-type CTNND1, which suggests a de novo mutation, but the possibility of low-abundance mutation in chimeric carriers cannot be ruled out. This mutation was not included in the dbSNP, 1,000 Genomes, or gnomAD databases, nor in the Human Gene Mutation Database or ClinVar database. According to the American College of Medical Genetics and Genomics guidelines, NM_001085458.2: c.566_c.567insG (p.Pro190fs*15) is a loss-of-function mutation, which may lead to loss of function (PVS1 level). This novel mutation (PS2 level) was confirmed using trio-based exome Sanger sequencing. The frequency of this mutation in the normal population is less than 0.0005 and has not been reported (PM2 level). Therefore, the NM_001085458.2: c.566_c.567insG (p.Pro190fs*15) mutation was classified as pathogenic, with the following rating evidence: PVS1 + PS2 + PM2. Sanger sequencing results confirmed the presence of this mutation in the fetus (Figure 2).
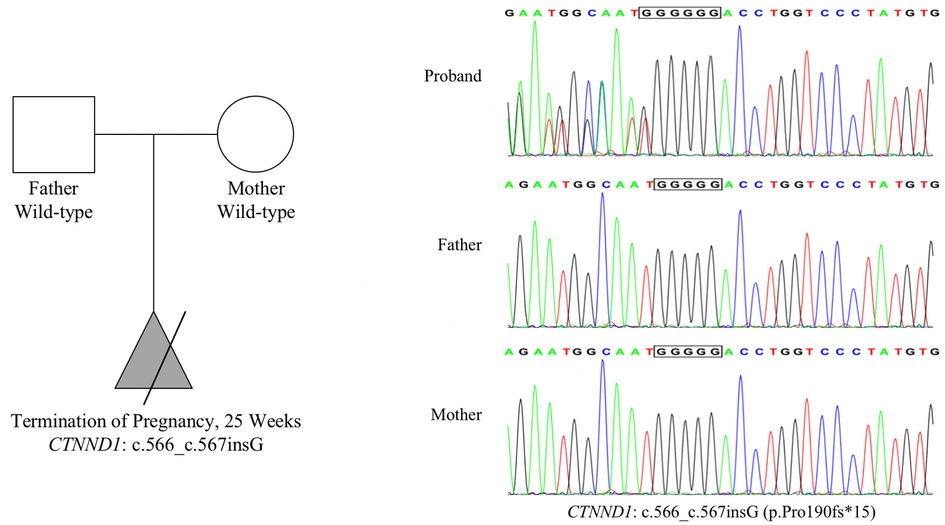
Figure 2. Mutation information. Whole-exome sequencing results show that the CTNND1 in the proband had a frameshift mutation: NM_001085458.2: c.566_c.567insG (p.Pro190fs*15). Both parents harbored wild-type CTNND1, suggesting a de novo mutation. Sanger sequencing confirmed this mutation.
Fetal CHD is one of the most common birth defects, with an incidence of approximately 4‰–5‰ in newborns (9). Single-gene mutations explain approximately 5% of CHD cases (9–11). In the present study, we report, to the best of our knowledge, the first instance of the prenatal diagnosis of major CHD, characterized by an apex-forming hypoplastic left ventricle, aortic coarctation, and a ventricular septal defect, in a fetus with a novel CTNND1 mutation (p.Pro190fs*15) that could be etiologic.
CTNND1 is located on chromosome 11q12.1, consists of 21 exons, and generates 22 transcripts (https://www.ncbi.nlm.nih.gov/). Its encoded protein, p120-ctn, is an Armadillo-repeat protein that interacts with the intracellular domain of E-cadherin, stabilizes the trans-membrane domain of E-cadherin, and plays a role in adhesion and signal transduction between cells (12). In Xenopus laevis, Ctnnd1 is highly expressed in the craniofacial bones and epidermis. In addition, p120-ctn plays an important role in the development of mouse eyelids and teeth (6, 13, 14). The main manifestations of human BCDS2 caused by CTNND1 defects are oral abnormalities, including congenital tooth hypoplasia, retention of deciduous teeth, and delayed eruption of permanent teeth. Abnormal tooth morphology includes small/nail-shaped lateral incisors in permanent dentition and crown cracks in lateral incisors. Eyelid malformations are also characteristic phenotypes and include lower eyelid ectropion, upper eyelid deformity, and ptosis. In addition, some patients present developmental retardation, language retardation, and partial hypoplasia of the genitourinary system and corpus callosum (3, 7, 15, 16). Therefore, CTNND1 is likely involved in different functions.
According to a previous study, BCDS mainly manifests as craniofacial malformations and oral abnormalities (3). However, subsequent studies have shown that CTNND1 might be associated with CHD. In the BCDS cohort reported by Alharatani et al., the incidence of CHD was approximately 46% (6/13), including tetralogy of Fallot, aortic arch hypoplasia, aortic stenosis, ventricular septal defects, atrial septal defects, patent ductus arteriosus, and patent foramen ovale (7). Although the role of CTNND1 in structural cardiac malformation remains unclear, it may be related to the development of the neural crest, which plays a crucial role in that of the cardiac diaphragm and valves (17). Indeed, Alharatani et al. showed that p120-ctn is highly expressed in human, mouse, and Xenopus embryonic hearts, and a functional defect in this protein might cause a cardiac phenotype by interfering with Wnt signal transduction in the neural crest (7).
To date, only 15 CTNND1 mutations have been identified in 19 patients. The main types are frameshift and nonsense mutations, with only one splice mutation (Table 1). These mutations are all loss-of-function mutations and may produce a truncated p120-ctn protein. There seems to be no correlation between a CTNND1 genotype and phenotype, which may be explained by the reduced number of reported patients with BCDS caused by CTNND1 defects and the inability to screen them. However, Alharatani et al. observed that if the truncation mutation is located at the C-terminal of p120-ctn, complete cleft lip and cleft palate may appear, as opposed to the phenotype when its location is at the N-terminal regulatory region; therefore, they speculated that truncation mutations at the p120-ctn C-terminal cause a dominant negative effect (7). In agreement with the latter, the CTNND1 p.Pro190fs*15 mutation reported in this study is located at the p120-ctn N-terminal regulatory region, and the fetus did not show a cleft lip or palate.
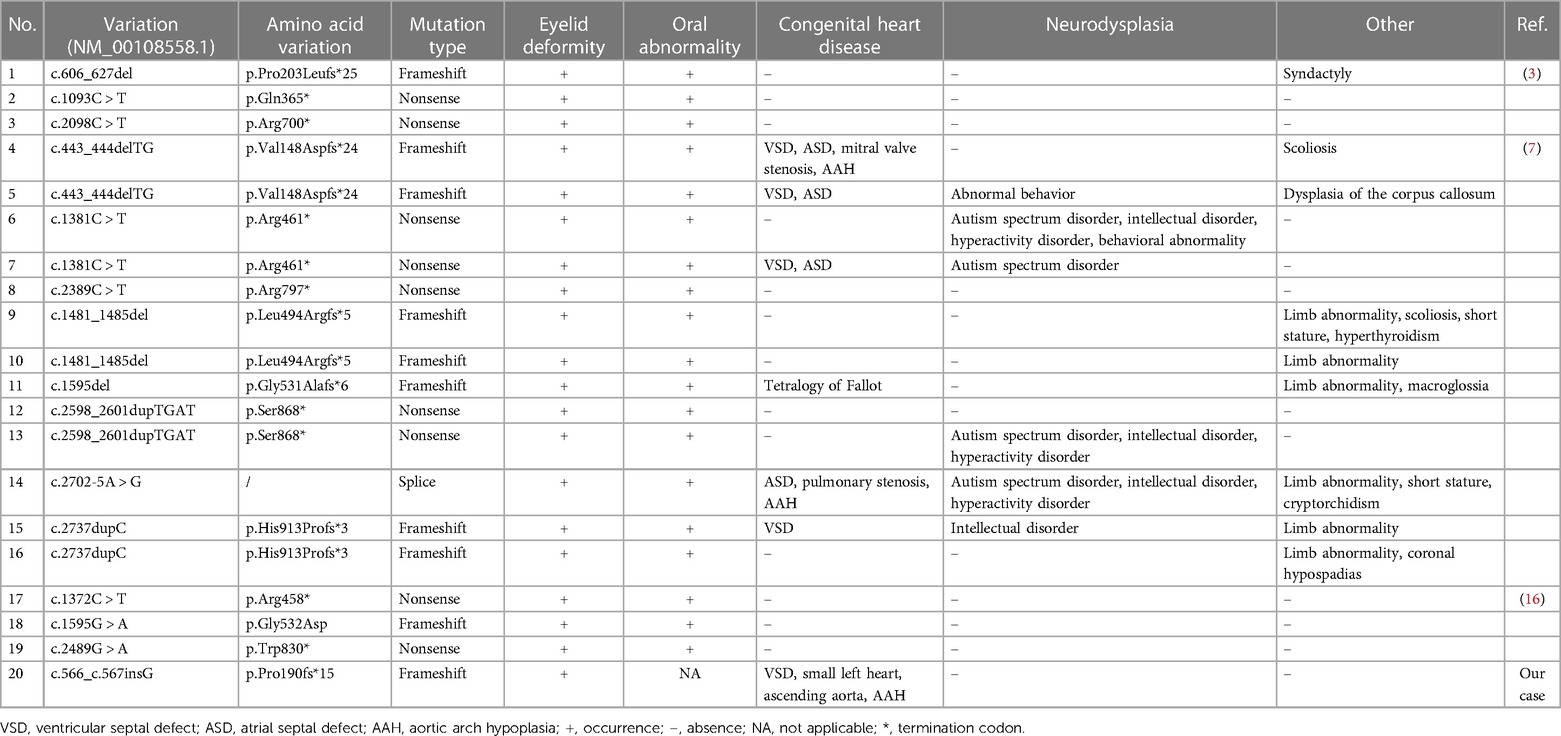
Table 1. Clinical phenotypes of patients harboring the CTNND1 mutation.
In summary, we report a prenatal case of complex cardiac structural abnormalities at 24 + 3 weeks of gestation. Whole-exome sequencing and Sanger sequencing were performed using fetal tissues after labor induction. The results showed a novel CTNND1 frameshift mutation, which may explain the early-onset CHD. We suggest CTNND1 p.Pro190fs*15 as a pathogenic factor leading to fetal CHD, especially in the absence of typical craniofacial malformations or abnormal oral phenotypes. Our findings broaden the CTNND1 mutation and phenotype spectra and reinforce the need for genetic testing at prenatal diagnosis in the face of complex cardiac phenotypes to improve diagnosis rates and genetic counseling during reproduction.
The original data presented in the study are included in the article material; further inquiries can be directed to the corresponding author. The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: ClinVar database repository, accession number SCV003845184 (https://www.ncbi.nlm.nih.gov/clinvar/).
The studies involving human participants were reviewed and approved by The 901th Hospital of the Joint Service of the People's Liberation Army. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
XZ, ZW, MY, and MZ performed the molecular genetic experiments. XZ, XL, ZW, MY, and MZ prepared the clinical and imaging data. WS performed gene and gene database analysis. XZ, ZW, and RT wrote the first draft of the manuscript. All authors contributed to the article and approved the submitted version.
This study was funded by the Research Fund Project of Anhui Medical University (ID: 2022xkj128).
The authors are grateful to the patients and their families for participating in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ababneh FK, Al-Swaid A, Elhag A, Youssef T, Alsaif S. Blepharo-cheilo-dontic (BCD) syndrome: expanding the phenotype, case report and review of literature. Am J Med Genet A. (2014) 164A(6):1525–9. doi: 10.1002/ajmg.a.36465
2. Weaver KN, Rutledge KD, Grant JH, Robin NH. Imperforate anus is a rare associated finding in blepharocheilodontic syndrome. Am J Med Genet A. (2010) 152A(2):438–40. doi: 10.1002/ajmg.a.33207
3. Ghoumid J, Stichelbout M, Jourdain A-S, Frenois F, Lejeune-Dumoulin S, Alex-Cordier M-P, et al. Blepharocheilodontic syndrome is a CDH1 pathway-related disorder due to mutations in CDH1 and CTNND1. Genet Med. (2017) 19(9):1013–21. doi: 10.1038/gim.2017.11
4. Oas RG, Xiao K, Summers S, Wittich KB, Chiasson CM, Martin WD, et al. p120-Catenin is required for mouse vascular development. Circ Res. (2010) 106(5):941–51. doi: 10.1161/CIRCRESAHA.109.207753
5. Marciano DK, Brakeman PR, Lee C-Z, Spivak N, Eastburn DJ, Bryant DM, et al. P120 catenin is required for normal renal tubulogenesis and glomerulogenesis. Development. (2011) 138(10):2099–109. doi: 10.1242/dev.056564
6. Bartlett JD, Dobeck JM, Tye CE, Perez-Moreno M, Stokes N, Reynolds AB, et al. Targeted p120-catenin ablation disrupts dental enamel development. PLoS One. (2010) 5(9):e12703. doi: 10.1371/journal.pone.0012703
7. Alharatani R, Ververi A, Beleza-Meireles A, Ji W, Mis E, Patterson QT, et al. Novel truncating mutations in CTNND1 cause a dominant craniofacial and cardiac syndrome. Hum Mol Genet. (2020) 29(11):1900–21. doi: 10.1093/hmg/ddaa050
8. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
9. van der Bom T, Zomer AC, Zwinderman AH, Meijboom FJ, Bouma BJ, Mulder BJM. The changing epidemiology of congenital heart disease. Nat Rev Cardiol. (2011) 8(1):50–60. doi: 10.1038/nrcardio.2010.166
10. Russell MW, Chung WK, Kaltman JR, Miller TA. Advances in the understanding of the genetic determinants of congenital heart disease and their impact on clinical outcomes. J Am Heart Assoc. (2018) 7(6):e006906. doi: 10.1161/JAHA.117.006906
11. Hopkins MK, Dugoff L, Kuller JA. Congenital heart disease: prenatal diagnosis and genetic associations. Obstet Gynecol Surv. (2019) 74(8):497–503. doi: 10.1097/OGX.0000000000000702
12. Cadwell CM, Su W, Kowalczyk AP. Cadherin tales: regulation of cadherin function by endocytic membrane trafficking. Traffic. (2016) 17(12):1262–71. doi: 10.1111/tra.12448
13. Ciesiolka M, Delvaeye M, Van Imschoot G, Verschuere V, McCrea P, van Roy F, et al. P120 catenin is required for morphogenetic movements involved in the formation of the eyes and the craniofacial skeleton in Xenopus. J Cell Sci. (2004) 117(Pt 18):4325–39. doi: 10.1242/jcs.01298
14. Li C-Y, Cha W, Luder H-U, Charles R-P, McMahon M, Mitsiadis TA, et al. E-cadherin regulates the behavior and fate of epithelial stem cells and their progeny in the mouse incisor. Dev Biol. (2012) 366(2):357–66. doi: 10.1016/j.ydbio.2012.03.012
15. Lin B, Liu Y, Su L, Liu H, Feng H, Yu M, et al. A novel CDH1 variant identified in a Chinese family with blepharocheilodontic syndrome. Diagnostics (Basel). (2022) 12(12):2936. doi: 10.3390/diagnostics12122936
16. Kievit A, Tessadori F, Douben H, Jordens I, Maurice M, Hoogeboom J, et al. Variants in members of the cadherin-catenin complex, CDH1 and CTNND1, cause blepharocheilodontic syndrome. Eur J Hum Genet. (2018) 26(2):210–9. doi: 10.1038/s41431-017-0010-5
Keywords: blepharocheilodontic syndrome, mutation, CTNND1, congenital heart disease, prenatal diagnosis
Citation: Zhao X, Li X, Sun W, Wei Z, Yu M, Zhang M and Tian R (2023) Case report: “Major fetal cardiac pathology associated with a novel CTNND1 mutation”. Front. Pediatr. 11:1180381. doi: 10.3389/fped.2023.1180381
Received: 6 March 2023; Accepted: 2 May 2023;
Published: 19 May 2023.
Edited by:
Lisa K. Hornberger, University of Alberta, CanadaReviewed by:
Birsen Karaman, Istanbul University, Türkiye© 2023 Zhao, Li, Sun, Wei, Yu, Zhang and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruixia Tian dGlhbnJ1aXhpYV8wMUAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.