 Hongyun Zhang1
Hongyun Zhang1 Yanling Teng
Yanling Teng Desheng Liang
Desheng Liang Zhuo Li
Zhuo Li Lingqian Wu
Lingqian Wu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 01 August 2023
Sec. Genetics of Common and Rare Diseases
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1177137
The prenatal prevalence of isolated ventriculomegaly is 0.039%–0.087%. Most isolated mild ventriculomegaly (MV) fetuses (>90%) have a favorable prognosis. However, 5.6% to 7.9% of fetuses with isolated MV have adverse neurodevelopmental outcomes. In this study, we reported the first case of prenatal Snijders Blok-Fisher syndrome (OMIM: #618604) caused by a truncating variant of POU3F3 (OMIM: *602480) in a fetus with transient isolated bilateral MV. The results of karyotype analysis, chromosomal microarray analysis, and TORCH infection evaluation for the fetus were all negative. However, a de novo likely pathogenic nonsense variant of NM_006236.3 (POU3F3): c.640C > T [rs1254251078] p.(Q214*) was identified by whole-exome sequencing (WES). Despite sufficient genetic counseling, the mother refused to undertake further brain magnetic resonance imaging (MRI) and decided to keep the fetus. She gave birth to a male infant through a full-term vaginal delivery. With a long-term follow-up, the infant unfortunately gradually presented with delayed motor development. The postnatal brain MRI of the proband showed dysplasia of the corpus callosum and ventriculomegaly. Considering the high probability of misdiagnosis for such cases, we further summarized the prenatal phenotypes from 19 reported patients with variants in POU3F3. The results revealed that 14 patients displayed a normal prenatal ultrasonographic manifestation, while only approximately 26.32% of fetuses showed MV or cysts without structural deformity. Thus our findings expand the variant spectrum of POU3F3 and suggest the importance of undertaking WES and brain MRI when the fetus has isolated bilateral MV.
Ventriculomegaly is one of the most common findings in prenatal ultrasounds, with a prevalence of 0.3–1.5 per 1,000 live births (1). Isolated ventriculomegaly is considered when no additional sonographic or chromosomal aberrations are identified, with a prenatal prevalence of 0.039%–0.087% (2, 3). When the diameter of the atrium of the lateral ventricles reaches 10–15 mm, it can be classified as mild lateral ventriculomegaly (MV), with a prenatal prevalence of 0.07% (3, 4). Most isolated mild ventriculomegaly (MV) fetuses (>90%) have a favorable prognosis. In contrast, an adverse neurodevelopmental outcome in fetal isolated MV can account for 5.6% to 7.9% of cases (3). Hence, for those fetuses with isolated MV, continuous ultrasound monitoring, brain magnetic resonance imaging (MRI), TORCH infection evaluation, and genetic testing (2) are suggested to explore the potential etiology to facilitate pregnancy management and predict neurodevelopmental outcomes.
In 2019, Petrovski et al. identified a POU3F3 heterozygous missense variant in a fetus with ventriculomegaly (5). POU3F3 (POU domain, class 3, transcription factor 3) is a member of the class III POU family of transcription factors, which is essential to cortical neuronal migration and neurogenesis (6). The variants in POU3F3 (OMIM: *602480) can cause an autosomal dominant neurodevelopmental disorder named Snijders Blok-Fisher syndrome (SBS). SBS (OMIM: #618604) is mainly manifested by global developmental delay and intellectual disability. A few patients may have epilepsy. The human gene mutation database (HGMD) has recorded 23 variants of POU3F3 up to February 2023. However, the truncating variants in POU3F3 have not been associated with ventriculomegaly.
This report described a fetus with a truncating de novo likely pathogenic variant of POU3F3 verified by trio whole-exome sequencing (WES) with transient isolated bilateral MV. The fetal results of karyotype analysis, chromosomal microarray analysis (CMA), and TORCH infection evaluation were negative. For the first time, this report associates a truncating variant in POU3F3 with the transient isolated bilateral MV.
In the present study, we identified a novel de novo POU3F3 nonsense variant in a Chinese fetus with isolated bilateral MV. The mother and father were 27 and 31 years old, respectively. There was no history of exposure to toxic substances and radiation during preconception planning and pregnancy. Regular maternity checkups were undertaken without abnormalities. There was no abnormal abdominal pain or vaginal bleeding during pregnancy. The results of non-invasive prenatal testing, nuchal translucency, and TORCH infection evaluation were negative. Amniocentesis was performed due to bilateral MV (left: 11.3 mm, and right: 10.9 mm), and a right choroid plexus cyst in the fetus was found by ultrasound at 24 weeks of gestation age (Figure 1A).
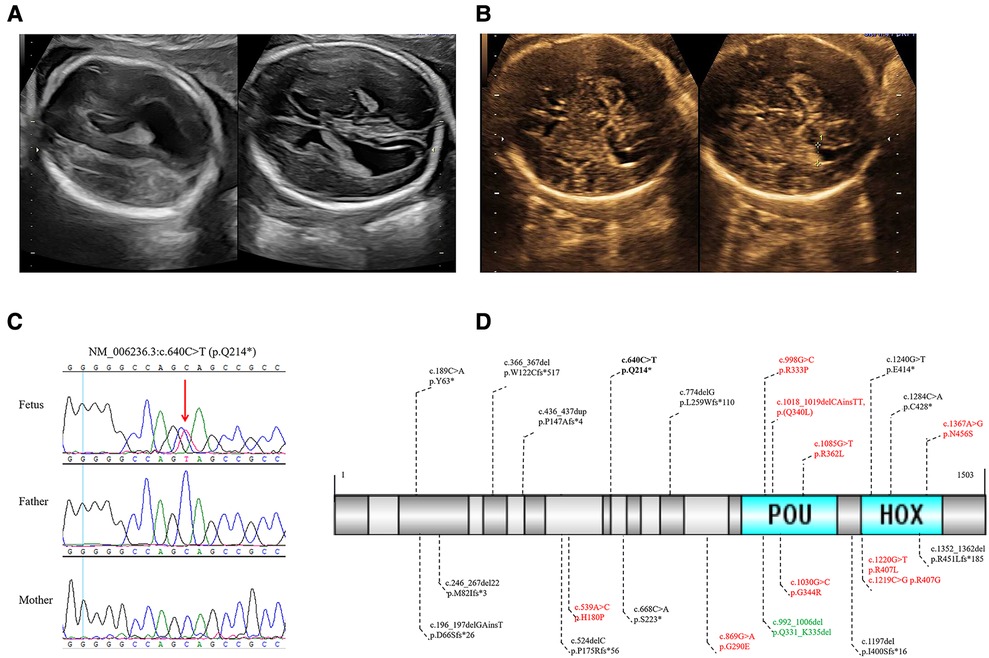
Figure 1. (A) The 24 weeks ultrasound of the fetus showed bilateral MV. (B) The 32 weeks ultrasound of the fetus showed a normal high value of bilateral ventricles. (C) Sanger sequencing validated the novel de novo likely pathogenic variant of POU3F3 in the fetus and healthy parents. (D) In all, 23 POU3F3 variants were recorded in HGMD up to February 2023. One variant was the gross deletion of the entire POU3F3, which is not in (D). Black, truncating variants; bold black, the variant in this study; red, missense variants; green, inframe deletion.
The amniotic fluid cells were collected to undertake the karyotype analysis, CMA, and WES, of which WES was performed in the fetus and healthy parents. The results of karyotyping and CMA were negative. The average depth in WES was 107X. The variants were analyzed in inheritance patterns, genotype-phenotype associations, and the pathogenicity evaluation after paired-end sequencing, alignment, variant calling, and annotation. We identified a novel de novo nonsense variant of NM_006236.3 (POU3F3): c.640C > T [rs1254251078] p.(Q214*) through trio WES (Figures 1C, Supplementary Figure S1). This variant was neither found in 1000G nor GnomAD; the CADD score was 36. Seven algorithms predicted that the variant was harmful (Supplementary Table S1). The final pathogenicity of c.640C > T was a likely pathogenic variant (PVS1_strong, PS2_moderate, and PM2_supporting) according to the American College of Medical Genetics and Genomics (ACMG) guidelines. The Sanger sequencing verified the likely pathogenic variant found by WES (Figure 1C). After consultation, the laboratory technician, genetic consultant, and clinician suggested that the parents undertake a brain MRI and pregnancy management.
During the follow-up, fetal ultrasound (32 weeks) showed a normal high value of bilateral ventricles (left: 9.5 mm, and right: 9.8 mm) and a right choroid plexus cyst (Figure 1B). The mother refused to undertake a brain MRI and gave birth to a full-term vaginal delivery of a male infant. No abnormality at birth and delivery was noted, and neonatal screening was negative. At the last visit, the child was two years old. He could sit at seven months old and walk with support after rehabilitation training at 1.5 years old. He had facial dysmorphism (ocular hypertelorism, protruding ears, and esotropia) and delayed fine and gross motor function. Moreover, the proband had mild developmental delays. However, he did not have speech delay, epilepsy, sleep disorder, autism, attention deficit hyperactivity disorder, joint hypermobility, and scoliosis. Postnatal brain MRI of the proband showed dysplasia of the corpus callosum and ventriculomegaly. Unfortunately, his parents were not willing to provide his brain MRI images.
We summarized the phenotypes from 48 patients with variants in POU3F3 (Table 1). In all, 44 (91.67%) patients had developmental delays and/or intellectual disabilities, and the clinical information of the other four patients was unknown. There was a phenotype difference between the non-truncating and truncating variants. For example, epilepsy was more common in patients with a non-truncating variant of POU3F3 (4/17, 23.53%), while only one patient had epilepsy in 27 patients with a truncating variant of POU3F3 (1/27, 3.70%). Moreover, 14 of 19 fetuses with variants in POU3F3 had a normal prenatal ultrasound during pregnancy. Besides, abnormal prenatal ultrasound of fetuses (5/19, 26.32%) mostly showed MV or cysts without structural deformity (5–7). In contrast, 10 of 22 (45.45%) patients had an abnormal postnatal brain MRI.
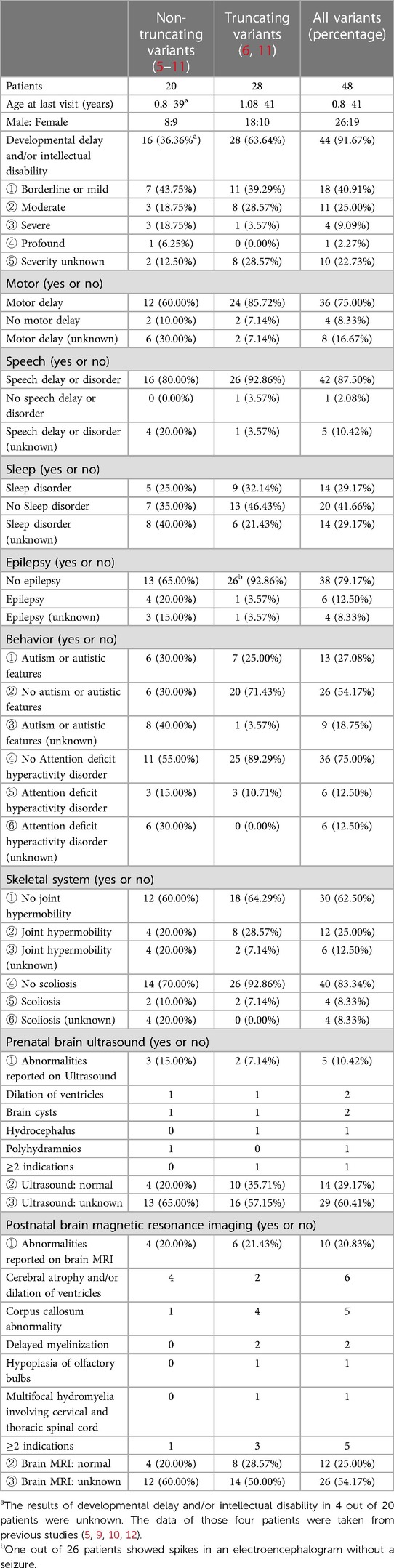
Table 1. Summary of phenotypes in 48 individuals with POU3F3 variants.
Up to February 2023, the HGMD had recorded 23 variants in POU3F3, of which 54.55% were truncating variants (Figure 1D). The penetrance of reported microdeletions or duplications in POU3F3 is of high penetrance (12). The post-transcriptional monitoring mechanism of nonsense-mediated mRNA decay (NMD) helps impede the translation of abnormally truncated proteins (13). However, POU3F3 is an intronless gene, which may lead to NMD escape and the impossibility to apply the PVS1 criterion of the ACMG/AMP pathogenicity classification. c.196_197delinsT (p.D66Sfs*26), c.668C > A (p.S223*), and c.1197delG (p.I400Sfs*16) were around c.640C > T (p.Q214*) in POU3F3 (Figure 1D). Snijders Blok et al. found that NMD escape was found in HEK293 cells with c.196_197delinsT (p.D66Sfs*26), c.668C > A (p.S223*), or c.1197delG (p.I400Sfs*16). The pathogenic mechanisms of c.196_197delinsT, c.668C > A, and c.1197delG were aberrant cytoplasmic expression and the decrease of dimerization capacity (6), which indicated the truncated regions in c.196_197delinsT, c.640C > T, c.668C > A, and c.1197delG were critical to the function of POU3F3. Besides, the pathogenic mechanism of POU3F3 was loss-of-function, which made it possible to apply the PVS1_strong criterion in the pathogenicity evaluation of c.640C > T, according to ACMG guidelines (14).
There is a difference in the proportion of prenatal (5/19, 26.32%) and postnatal (10/22, 45.45%) brain abnormalities among patients with variants in POU3F3. Two possible reasons may cause this phenomenon. First, ultrasounds have a lower resolution in detecting brain tissue than MRIs, which can lead to fine structural deformities not being diagnosed by ultrasound (15, 16). Second, the patients with variants in POU3F3 may not have brain structural deformity at birth, and the postnatal brain structural deformity may be progressive.
Most isolated MV fetuses (>90%) have a favorable prognosis. However, 5.6% to 7.9% of fetuses with isolated MV have adverse neurodevelopmental outcomes (3). Therefore, it is essential to undertake continuous ultrasound monitoring, genetic testing, brain MRIs, and TORCH infection evaluation for a comprehensive evaluation of fetal MV under the current guideline (2, 17). Some studies have found that MV has recently been associated with monogenic disorders (18, 19). For example, MV is found to be the most common prenatal ultrasound phenotype in MDS/PAFAH1B1-related lissencephaly (18). Moreover, the diagnostic yield of fetal isolated MV is 13% (3/23) through WES (20). According to the guideline of fetal MV from the Society for Maternal-Fetal Medicine in 2018 (17), these fetuses with isolated MV caused by monogenic disorders would have been missed diagnoses. Therefore, we recommended that WES should be the last diagnostic option for fetuses with isolated bilateral MV but a normal CMA result. Exploring the genetic etiology of isolated MV facilitates pregnancy management and predicts neurodevelopmental outcomes, and applying WES and brain MRI promotes fetal intrauterine phenotype-genotype association enrichment.
In conclusion, we report the first case of prenatal Snijders-Blok-Fisher syndrome caused by a de novo heterozygous truncating variant of POU3F3 in a fetus with transient isolated bilateral MV. It suggests the importance of undertaking WES and brain MRI when the fetus has isolated bilateral MV. Exploring the genetic etiology of isolated MV facilitates pregnancy management and predicts neurodevelopmental outcomes. Besides, our findings expand the variant spectrum and fetal intrauterine phenotype-genotype association of POU3F3.
The data presented in the study are deposited in the Genome Sequence Archive (GSA) (https://ngdc.cncb.ac.cn/gsa/) BioProject: PRJCA018171, Accession number HRA005034. Further requests can be directed to the corresponding author.
This study was conducted following the standards approved by the Ethics Committee of Central South University and the informed consent signed by the legal guardians of the fetus.
HZ: writing the manuscript, data collection, and genetic analysis; SLP and CP: recruitment of the family members; YT: genetic analysis; DL: investigation; and ZL and LW: funding acquisition and supervision. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Key R&D Program of China (2021YFC1005303, 2021YFC1005302, 2021YFC1005301), the National Natural Science Foundation of China (81970829, 81974240, 82171711), the Science Technology Innovation Program of Hunan Province (2019SK1010, 2019SK1014), and the Key R&D Program of Zhejiang Province of China (2021C03030).
We thank the proband's parents for their support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2023.1177137/full#supplementary-material
1. Mehlhorn AJ, Morin CE, Wong-You-Cheong JJ, Contag SA. Mild fetal cerebral ventriculomegaly: prevalence, characteristics, and utility of ancillary testing in cases presenting to a tertiary referral center. Prenat Diagn. (2017) 37(7):647–57. doi: 10.1002/pd.5057
2. Scelsa B, Rustico M, Righini A, Parazzini C, Balestriero MA, Introvini P, et al. Mild ventriculomegaly from fetal consultation to neurodevelopmental assessment: a single center experience and review of the literature. Eur J Paediatr Neurol. (2018) 22(6):919–28. doi: 10.1016/j.ejpn.2018.04.001
3. Thorup E, Jensen LN, Bak GS, Ekelund CK, Greisen G, Jørgensen DS, et al. Neurodevelopmental disorder in children believed to have isolated mild ventriculomegaly prenatally. Ultrasound Obstet Gynecol. (2019) 54(2):182–9. doi: 10.1002/uog.20111
4. Pagani G, Thilaganathan B, Prefumo F. Neurodevelopmental outcome in isolated mild fetal ventriculomegaly: systematic review and meta-analysis. Ultrasound Obstet Gynecol. (2014) 44(3):254–60. doi: 10.1002/uog.13364
5. Petrovski S, Aggarwal V, Giordano JL, Stosic M, Wou K, Bier L, et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet. (2019) 393(10173):758–67. doi: 10.1016/s0140-6736(18)32042-7
6. Snijders Blok L, Kleefstra T, Venselaar H, Maas S, Kroes HY, Lachmeijer AMA, et al. De Novo variants disturbing the transactivation capacity of POU3F3 cause a characteristic neurodevelopmental disorder. Am J Hum Genet. (2019) 105(2):403–12. doi: 10.1016/j.ajhg.2019.06.007
7. Torun D, Arslan M. Coexistence of severe developmental delay, epilepsy, and hemangioma in snijders blok-fisher syndrome suggests the presence of a POU3F3-related SNIBFIS endophenotype: a case report. Am J Med Genet A. (2021) 185(5):1554–60. doi: 10.1002/ajmg.a.62135
8. Edwards JJ, Rouillard AD, Fernandez NF, Wang Z, Lachmann A, Shankaran SS, et al. Systems analysis implicates WAVE2 Complex in the pathogenesis of developmental left-sided obstructive heart defects. JACC Basic Transl Sci. (2020) 5(4):376–86. doi: 10.1016/j.jacbts.2020.01.012
9. Quaio C, Moreira CM, Novo-Filho GM, Sacramento-Bobotis PR, Groenner Penna M, Perazzio SF, et al. Diagnostic power and clinical impact of exome sequencing in a cohort of 500 patients with rare diseases. Am J Med Genet C Semin Med Genet. (2020) 184(4):955–64. doi: 10.1002/ajmg.c.31860
10. Sparks TN, Lianoglou BR, Adami RR, Pluym ID. Exome sequencing for prenatal diagnosis in nonimmune hydrops Fetalis. N Engl J Med. (2020) 383(18):1746–56. doi: 10.1056/NEJMoa2023643
11. Rossi A, Blok LS, Neuser S, Klöckner C, Platzer K, Faivre LO, et al. POU3F3-related Disorder: defining the phenotype and expanding the molecular spectrum. Clin Genet. (2023) 104(2):186–97. doi: 10.1111/cge.14353.
12. Farwell Hagman KD, Shinde DN, Mroske C, Smith E, Radtke K, Shahmirzadi L, et al. Candidate-gene criteria for clinical reporting: diagnostic exome sequencing identifies altered candidate genes among 8% of patients with undiagnosed diseases. Genet Med. (2017) 19(2):224–35. doi: 10.1038/gim.2016.95
13. Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE. Nonsense-mediated decay approaches the clinic. Nat Genet. (2004) 36(8):801–8. doi: 10.1038/ng1403
14. Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. (2018) 39(11):1517–24. doi: 10.1002/humu.23626
15. Cooper S, Katorza E. Prenatal abnormal width of the cavum septum pellucidum - MRI features and neurodevelopmental outcome. J Matern Fetal Neonatal Med. (2018) 31(22):3043–50. doi: 10.1080/14767058.2017.1364721
16. Glenn OA, Goldstein RB, Li KC, Young SJ, Norton ME, Busse RF, et al. Fetal magnetic resonance imaging in the evaluation of fetuses referred for sonographically suspected abnormalities of the corpus callosum. J Ultrasound Med. (2005) 24(6):791–804. doi: 10.7863/jum.2005.24.6.791
17. Fox NS, Monteagudo A, Kuller JA, Craigo S, Norton ME. Mild fetal ventriculomegaly: diagnosis, evaluation, and management. Am J Obstet Gynecol. (2018) 219(1):B2–9. doi: 10.1016/j.ajog.2018.04.039
18. Zhang YL, Jing XY, Zhen L, Pan M, Han J, Li DZ. Prenatal diagnosis of miller-dieker syndrome/PAFAH1B1-related lissencephaly: ultrasonography and genetically investigative results. Eur J Obstet Gynecol Reprod Biol. (2022) 274:28–32. doi: 10.1016/j.ejogrb.2022.04.025
19. Gao S, Zhao X, Zhao G, Dai P, Kong X. Analysis of L1CAM gene mutation and imaging appearance in three Chinese families with L1 syndrome: three case reports. Mol Genet Genomic Med. (2022) 10(9):e2002. doi: 10.1002/mgg3.2002
Keywords: transient isolated bilateral mild ventriculomegaly, POU3F3, whole-exome sequencing, snijders blok-Fisher syndrome, truncating variant
Citation: Zhang H, Linpeng S, Teng Y, Peng C, Liang D, Li Z and Wu L (2023) A de novo heterozygous POU3F3 genotype for the p.(Q214*) variant in a fetus with transient isolated bilateral mild ventriculomegaly: a case report and review of the literature. Front. Pediatr. 11:1177137. doi: 10.3389/fped.2023.1177137
Received: 14 April 2023; Accepted: 13 July 2023;
Published: 1 August 2023.
Edited by:
Ting Hu, Sichuan University, ChinaReviewed by:
Miguel Angel Alcántara-Ortigoza, National Institute of Pediatrics, Mexico© 2023 Zhang, Linpeng, Teng, Peng, Liang, Li and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhuo Li bGl6aHVvQHNrbG1nLmVkdS5jbg== Lingqian Wu d3VsaW5ncWlhbkBza2xtZy5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.