 Mattia Pozzato1,2,†
Mattia Pozzato1,2,† Robertino Dilena3,†
Robertino Dilena3,† Greta Rogani4Gisella Beretta4
Greta Rogani4Gisella Beretta4 Sofia Torreggiani4Stefano Lanni4Alessandra Tozzo5Francesca Andreetta6
Sofia Torreggiani4Stefano Lanni4Alessandra Tozzo5Francesca Andreetta6 Paola Cavalcante6
Paola Cavalcante6 Fabio Triulzi7
Fabio Triulzi7 Filippo Martinelli Boneschi8
Filippo Martinelli Boneschi8 Francesca Minoia4
Francesca Minoia4 Giovanni Filocamo4*
Giovanni Filocamo4*
- 1Neurology Unit & MS Centre, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy
- 2Dino Ferrari Centre, Neuroscience Section, Department of Pathophysiology and Transplantation, University of Milan, Milan, Italy
- 3Neuropathophysiology Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy
- 4Pediatric Immunorheumatology Unit, Fondazione IRCCS Ca' Granda ospedale Maggiore Policlinico, Milan, Italy
- 5Infantile Neuropsychiatry Unit, Pediatric Neuroscience Department, IRCCS Fondazione Istituto Neurologico “C. Besta”, Milan, Italy
- 6Neurology 4 - Neuroimmunology and Neuromuscular Diseases Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy
- 7Neuroradiology Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy
- 8Clinical Neurology, Department of Health Science CRC “Aldo Ravelli” for Experimental Brain Therapeutics, Hospital San Paolo ASST Santi Paolo e Carlo Milan and University of Milan, Milan, Italy
Behcet's disease (BD) is a rare vasculitis characterized by multisystemic inflammation. Central nervous system (CNS) involvement is rare and heterogeneous, particularly in the pediatric population. A diagnosis of neuro-Behcet could be highly challenging, especially if neurological manifestations precede other systemic features; however, its timely definition is crucial to prevent long-term sequelae. In this study, we describe the case of a girl who, at 13 months of age, presented with a first episode of encephalopathy compatible with acute disseminated encephalomyelitis, followed, after 6 months, by a neurological relapse characterized by ophthalmoparesis and gait ataxia, in association with new inflammatory lesions in the brain and spinal cord, suggesting a neuromyelitis optica spectrum disorder. The neurological manifestations were successfully treated with high-dose steroids and intravenous immunoglobulins. In the following months, the patient developed a multisystemic involvement suggestive of Behcet's disease, characterized by polyarthritis and uveitis, associated with HLA-B51 positivity. The challenge presented by this unique case required a multidisciplinary approach involving pediatric neurologists, neuro-radiologists, and pediatric rheumatologists, with all of these specialists creating awareness about early-onset acquired demyelinating syndromes (ADSs). Given the rarity of this presentation, we performed a review of the literature focusing on neurological manifestations in BD and differential diagnosis of patients with early-onset ADS.
Introduction
Behcet's disease (BD) is an inflammatory vasculitis affecting the arteries or veins of all sizes and is characterized by cutaneous, ocular, articular, gastrointestinal, and/or central nervous system involvement. The first symptoms and signs of the disease commonly appear between the second and the fourth decade of life, but it can rarely develop in childhood and even in infancy (1).
Neurological involvement in pediatric BD is rare, but its early diagnosis is essential to prevent unfavorable outcomes. Central nervous system (CNS) manifestations can be attributed to two major pathways: first, and most commonly, through the development of an immune-mediated meningoencephalitis, which predominantly involves the brainstem, but can also involve the basal ganglia, thalamus, cortex, white matter, spinal cord, or cranial nerves; and second, as a consequence of thrombosis within the dural venous sinuses (2).
In this study, we describe the case of a very young girl who, at 13 months of age, presented the first neurological manifestation with radiologic features of acute disseminated encephalomyelitis (ADEM) and who subsequently developed a multisystemic involvement characterized by polyarthritis and uveitis suspicious for BD.
Case report
A 13-month-old girl presented with an asymmetric weakness and hypertonia in the four limbs (both arms and legs), which appeared more severe on the right side than on the left side, with right head deviation. She could only perform some abnormal dystonic movements with the right limbs because her left limbs were almost plegic. She also presented with an altered state of consciousness, alternating periods of sleepiness with severe psychomotor agitation. Notably, 4 days before her neurological symptoms occurred, she presented with fever and cough, and 1 month prior to this, the patient had received vaccination against diphtheria, tetanus, pertussis, poliomyelitis, haemophilus B, hepatitis B, and pneumococcus. A cerebrospinal fluid (CSF) analysis revealed that the level was within the normal range, and a broad screening for infectious agents showed a negative result. The patient was also tested for the presence of oligoclonal bands, anti-AQP4, and anti-MOG antibodies. Brain magnetic resonance imaging (MRI) showed symmetrical thalamic lesions, with extension to the hemispheric white matter and to the right optic tract, without contrast enhancement (Figure 1A). A diagnosis of acute disseminated encephalomyelitis (ADEM) was made, and endovenous high-dose methylprednisolone therapy was started for 7 days, which involved a cycle of high-dose intravenous immunoglobulins (IVIg), leading to rapid clinical recovery and radiological response at the 3-month control brain MRI, without any radiological progression (Figure 1B). Due to the unusual coexistence of extrapyramidal dystonic manifestations at onset, thiamine-responsive basal ganglia disease (BTBGD) was excluded by sequencing the SLC19A3 gene; likewise, inherited inborn errors of metabolism, including mitochondrial diseases, were excluded.
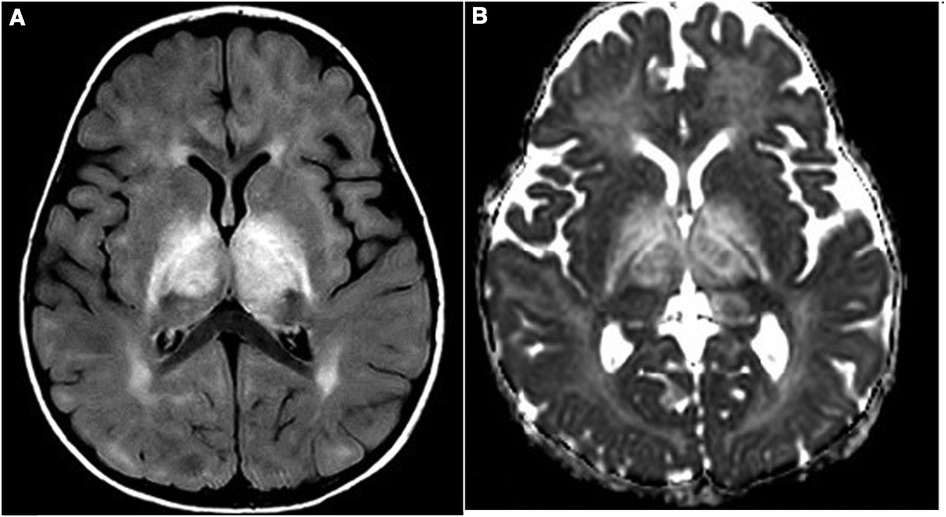
Figure 1. (A,B) First MRI study: FLAIR (A) and ADC (B) images show diffuse thalamic and internal capsule bilateral involvement with increased diffusion as for vasogenic edema.
Oral prednisone was tapered in 2 months. Four months later, the child reached the gait milestone, and a limping on the right leg was noticed, with an attitude in extra rotation of the right lower limb. One month later, and 17 days after the vaccination against measles, parotitis, and rubella, the patient presented with a second neurological episode characterized by the acute onset of gaze paralysis, left eye esophoria, vertical nystagmus, and inability to walk due to severe gait ataxia. The patient again underwent a brain and spinal cord MRI, which showed new lesions in the supra- and infratentorial white matter, optic nerves, some of which with gadolinium enhancement, and longitudinally extensive transverse myelitis (LTEM) lesion at the C2–C4 level, suggestive of neuromyelitis optica spectrum disorder (NMOSD) (Figure 2). Visual evoked potentials (EPs) showed an increase in P100 latency in both eyes, while brainstem auditory and somatosensory EP demonstrated an increased central conduction velocity. CSF analysis again showed that the level was within normal range, and no oligoclonal bands nor anti-AQP4 or anti-MOG antibodies were detected. However, a CSF cytokine panel assay showed multiple increases in proinflammatory cytokines, such as IL-6, IL-1Ra, MCP-1, and IP-10/CXCL10 (Supplementary Table S1). Blood tests showed antinuclear antibody (ANA) positivity (>1:640 titer) and the presence of HLA-B51. The patient was re-treated with high-dose steroids and monthly high-dose IVIg, which resulted in good neurological recovery, despite the persistence of a fluctuating limp in the right leg with a limited range of motion.
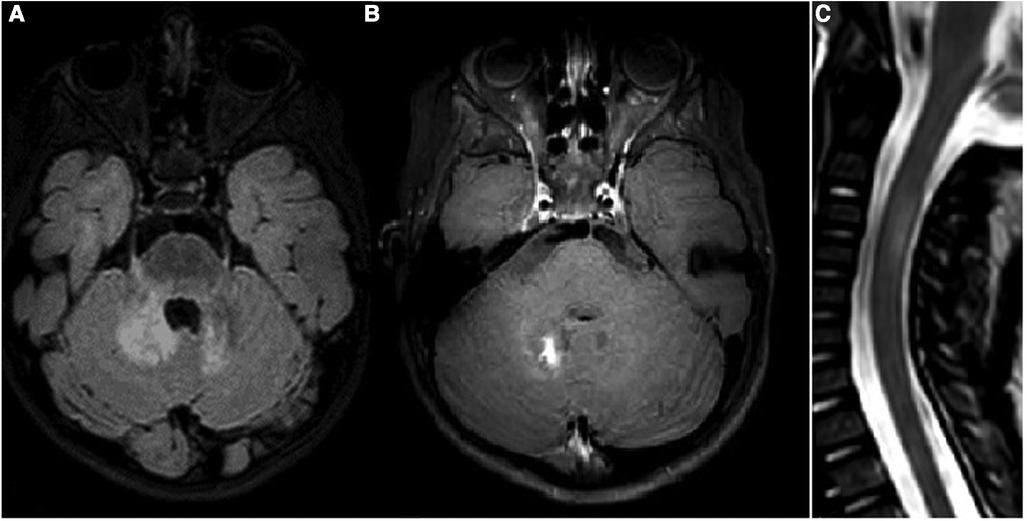
Figure 2. (A–C) Third MRI study: FLAIR (A) and postcontrast T1WI (B) of the cerebellum and a STIR midsagittal image of the cervical spine (C). New lesions on deep cerebellar white matter and V cranial nerves origin are visible, with a postcontrast enhancement of the right lesion. A new lesion in the cervical spine is also visible at the C3–C4 level.
An MRI performed at 29 months of age did not show any new brain or spinal cord lesion but highlighted the presence of active arthritis of the right temporomandibular joint (Supplementary Figure S1). A complete joint assessment performed at that time by pediatric rheumatologists revealed swelling, pain, and limitation in the motion of the right knee and ankle. The presence of active arthritis was confirmed by an ultrasound assessment.
The very early onset of neurological manifestations and the subsequent development of polyarthritis led us to consider the wide spectrum of inherited diseases characterized by neuroinflammation. Type I interferon signature was negative. Whole-exome sequencing was then performed, with a particular focus on the genes involved in neuroinflammatory pathways (3) without any evidence of pathogenic variants.
Given the broad differential diagnosis performed, the presence of CNS parenchymal lesions located in the brain and spinal cord with a relapsing-remitting course, the good clinical recovery with immunomodulating treatment, and the evidence of polyarthritis associated with HLA-B51 positivity suggested a possible diagnosis of Bechet's disease with neurological involvement.
Intra-articular steroid injections were administered on the right knee and ankle, and therapy with colchicine (0.5 mg/daily) and methotrexate (7.5 mg/weekly) was started, with a stabilization of the neurological picture both clinically and radiologically. Due to the occurrence of tibiotarsal arthritis relapses, two more intraarticular steroid injections with the addition of biologic therapy with anti-IL6 (tocilizumab) were required for the girl at the age of 3 years and 3 months, which resulted in a very good clinical response and complete articular remission.
However, 3 years and 6 months from the disease onset, the patient developed bilateral uveitis despite ongoing treatment with tocilizumab and was, therefore, switched to adalimumab as part of anti-TNFα treatment, following which she showed a positive clinical and ophthalmological response. At the last follow-up, performed at the age of 5 years, 6 months after the start of adalimumab, the patient appeared fine with the disease and uveitis in remission.
Discussion
Early-onset neurological manifestations are often a diagnostic challenge, especially when they are the first and isolated clinical features. In children, systemic symptoms may develop over the years and differential diagnosis covers a broad range of conditions, including systemic autoinflammatory/autoimmune disorders, acquired demyelinating syndromes (ADSs), and inherited autoinflammatory, neurological, and metabolic disorders. The case herein presented highlights this diagnostic dilemma.
ADSs represent a group of clinical conditions characterized by an inflammation of the CNS and consequent damage to the myelin. ADSs can be classified as follows: (1) monophasic forms, such as ADEM or clinically isolated syndrome (CIS); (2) polyphasic or chronic forms, including relapsing ADEM, multiple sclerosis (MS), NMOSD, or CNS involvement in the context of systemic inflammatory diseases, such as neuro-lupus, neuro-sarcoidosis, neuro-Bechet's, and others (4). A distinction needs to be made between monophasic demyelinating syndrome and polyphasic demyelinating syndrome, which is relevant for clinical management. In monophasic forms, steroids and IVIg are the treatments of choice, while chronic disorders often require disease-modifying therapy in order to prevent the accumulation of disability.
The complex workup in a pediatric patient with a suspicion of ADS is presented in Supplementary Figure S2), and the most common differential diagnoses are summarized and detailed in Table 1.
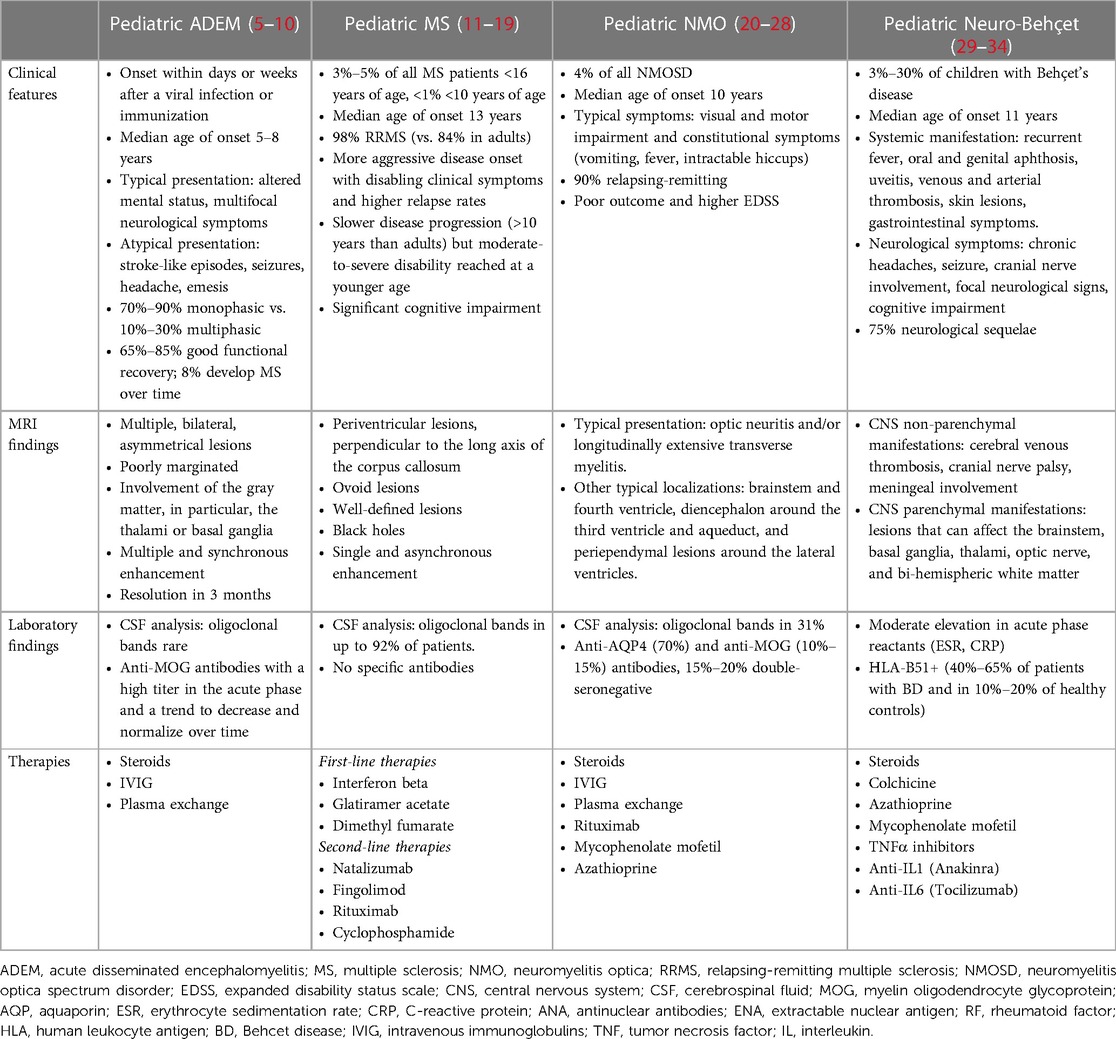
Table 1. Differential diagnosis in the context of acquired demyelinating syndromes in pediatric age with principal clinical characteristics.
Our patient had a very early-onset neurological manifestation, exceptionally described so far in BD (35). Aphthosis was never reported in our patient; however, oral aphthosis is rarely present during the first years of age, even in patients with a full-blown diagnosis of BD in older age (29, 30).
As no single pathognomonic test exists, BD still represents a clinical challenge, particularly in the pediatric population, and its diagnosis relies on clinical findings and the exclusion of mimicking conditions. In our patient, neuro systemic lupus erythematosus (SLE) was considered. However, the absence of any cutaneous, hematological, or renal signs of the disease, the absence of specific antibodies (double-strained DNA), extractable nuclear antigen (ENA), or antiphospholipids, and the absence of complement consumption did not support this hypothesis. Furthermore, the extremely young age of the patient would have been unusual for classic SLE. Despite the intriguing monogenic SLE/interpheronopathy, the interferon gene signature was negative and the whole-exome sequencing performed excluded all genes related to those conditions. The normal level of the angiotensin-converting enzyme (ACE) and the absence of granulomatous lesions, particularly of granulomatous uveitis, made neuro-sarcoidosis unlikely.
Adult criteria for BD showed poor sensitivity and/or specificity in children (31, 32). In 2015, a consensus classification criteria set, based on a cohort of 219 children with BD, was established for pediatric BD (PEDBD) (29). The diagnosis of BD can be considered in the presence of three of the six following recurrent symptoms: oral aphthous lesions, genital ulcers, skin involvement, eye involvement, neurological findings, and vascular findings. However, clinical symptoms might develop over the years in patients with Behcet’s disease, hampering the application of the classification criteria in daily clinical practice.
The term neuro-Behcet disease (NBD) is used to define the nervous system involvement in BD. The rate of pediatric NBD was variably reported in the literature from 3% to more than 30% of children (30, 33). Neurological manifestations can precede other systemic features for a long time, making the diagnostic workup challenging. Early-onset BD is often insidious, due to its proteiform presentation, and its prompt diagnosis, which is essential to prevent adverse outcomes, is difficult, particularly when a rare manifestation, such as the neurological one, is the leading or the only clinical feature.
NBD can be classified into two major forms: (1) “non-parenchymal or vascular” disease, characterized by intracranial hypertension, typically due to dural sinus thrombosis and (2) “parenchymal” disease, which may include recurrent encephalomyelitis, aseptic meningitis, cranial nerve palsy, and neuropathy. Although rare, cerebral venous thrombosis is the most common presentation of pediatric NBD (1). MRI findings, although aspecific, are essential to guide differential diagnosis (34). CSF usually reveals elevated protein levels and pleocytosis. Recent studies have suggested the involvement of proinflammatory pathways such as IL-1, IL-6, IL-33, and INFγ in the CSF of patients with NBD (36). HLA-B51 positivity is considered the most important genetic risk factor of the disease and is described in 40%–65% of patients with BD but also in 10%–20% of healthy subjects; therefore, it should not be considered a diagnostic criterium; however, its positivity during the occurrence of compatible symptoms may support the diagnosis of BD.
In a case series of 26 children with BD, 11 developed neurological abnormalities with neurologic involvement at a mean age of 13.5 years, although not all fulfilled the criteria of BD at the onset. Two patients presented with a neurologic attack prior to the systemic manifestations of BD. One had parenchymal CNS involvement as well as oral aphthae at the age of 8 and fulfilled the International Study Group for Behcet Disease (ISGBD) criteria only many years later (37). In another recent case series, five pediatric patients diagnosed with NBD were described. The mean age reported was 15 years and oral ulcers were present in all patients (38). Table 2 summarizes the prevalence and the neurological manifestations of pediatric patients with BD and neurological involvement reported in the literature.
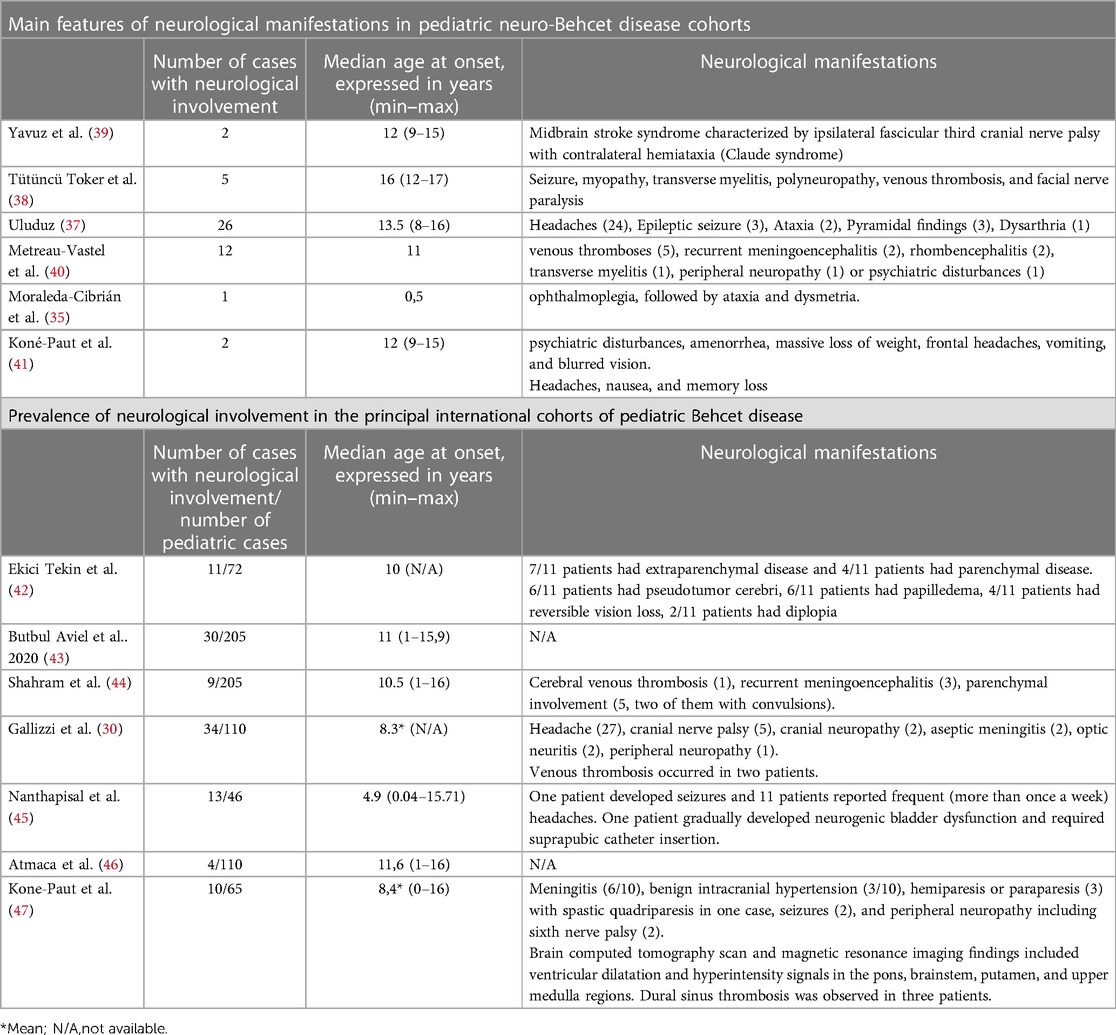
Table 2. Summary of the main features and prevalence of neurological involvement in the principal international cohorts of pediatric Behcet disease.
Conclusion
Diagnosis and treatment of pediatric NBD are challenging because of its rarity and heterogeneous presentation, particularly when neurological involvement occurs early in infancy and precedes systemic signs and symptoms. A multidisciplinary approach, involving neurologists, neuroradiologists, and rheumatologists, is crucial to define the most probable diagnosis in a timely manner and start an appropriate treatment regimen as soon as possible.
Early-onset NBD could severely affect psychomotor development, and therefore, a timely diagnosis is important to prevent the possible accumulation of disability and poor outcomes in adulthood.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors without undue reservation.
Ethics statement
Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
MP, RD, GR, and GB drafted the manuscript. FM and GF conceived the study’s design, conceptualized the study, and reviewed the manuscript. ST and SL collected the data. FT reviewed the MRI findings and also reviewed the manuscript. AT and FA performed the cytokine laboratory analysis, analyzed the data, and reviewed the manuscript. FM reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Publication [The APC] costs were funded by Grant Ricerca Corrente, Italian Ministry of Health.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2023.1175584/full#supplementary-material
SUPPLEMENTARY FIGURE S1 A–B
Fourth MRI study on TMJ; STIR sequence (a) and post contrast T1WI acquisition (B) show an inflammation of the right TMJ with slight bone edema on STIR (a, arrow) and periarticular enhancement after contrast (b, arrow).
SUPPLEMENTARY FIGURE S2
Flow-chart of the diagnostic approach to pediatric patients with a first neurological episode suggestive for demyelinating disease.
References
1. Mora P, Menozzi C, Orsoni JG, Rubino P, Ruffini L, Carta A. Neuro-Behçet’s disease in childhood: a focus on the neuro-ophthalmological features. Orphanet J Rare Dis. (2013) 8:18. doi: 10.1186/1750-1172-8-18
2. Al-Araji A, Kidd DP. Neuro-Behçet’s disease: epidemiology, clinical characteristics, and management. Lancet Neurol. (2009) 8(2):192–204. doi: 10.1016/S1474-4422(09)70015-8
3. McCreary D, Omoyinmi E, Hong Y, Mulhern C, Papadopoulou C, Casimir M, et al. Development and validation of a targeted next-generation sequencing gene panel for children with neuroinflammation. JAMA Netw Open. (2019) 2(10):e1914274. doi: 10.1001/jamanetworkopen.2019.14274
4. Carbonell CF, Chitnis T. Inflammatory demyelinating diseases in children: an update. Minerva Pediatr. (2013) 65(3):307–23.23685382
5. Alves-Leon SV, Veluttini-Pimentel ML, Gouveia ME, Malfetano FR, Gaspareto EL, Alvarenga MP, et al. Acute disseminated encephalomyelitis: clinical features, HLA DRB1*1501, HLA DRB1*1503, HLA DQA1*0102, HLA DQB1*0602, and HLA DPA1*0301 allelic association study. Arq Neuropsiquiatr. (2009) 67(3A):643–51. doi: 10.1590/S0004-282X2009000400013
6. Pohl D, Alper G, Van Haren K, Kornberg AJ, Lucchinetti CF, Tenembaum S, et al. Acute disseminated encephalomyelitis: updates on an inflammatory CNS syndrome. Neurology. (2016) 87(9 Supplement 2):S38–S45. doi: 10.1212/WNL.0000000000002825
7. Berzero G, Cortese A, Ravaglia S, Marchioni E. Diagnosis and therapy of acute disseminated encephalomyelitis and its variants. Expert Rev Neurother. (2016) 16(1):83–101. doi: 10.1586/14737175.2015.1126510
8. Tenembaum SN. Disseminated encephalomyelitis in children. Clin Neurol Neurosurg. (2008) 110(9):928–38. doi: 10.1016/j.clineuro.2007.12.018
9. Callen DJA, Shroff MM, Branson HM, Li DK, Lotze T, Stephens D, et al. Role of MRI in the differentiation of ADEM from MS in children. Neurology. (2009) 72(11):968–73. doi: 10.1212/01.wnl.0000338630.20412.45
10. Di Pauli F, Mader S, Rostasy K, Schanda K, Bajer-Kornek B, Ehling R, et al. Temporal dynamics of anti-MOG antibodies in CNS demyelinating diseases. Clin Immunol. (2011) 138(3):247–54. doi: 10.1016/j.clim.2010.11.013
11. Alroughani R, Boyko A. Pediatric multiple sclerosis: a review. BMC Neurol. (2018) 18(1):27. doi: 10.1186/s12883-018-1026-3
12. Boiko A, Vorobeychik G, Paty D, Devonshire V, Sadovnick D, the UBC MS Clinic Neurologists. Early onset multiple sclerosis: a longitudinal study. Neurology. (2002) 59(7):1006–10. doi: 10.1212/WNL.59.7.1006
13. Renoux C, Vukusic S, Mikaeloff Y, Edan G, Clanet M, Dubois B, et al. Natural history of multiple sclerosis with childhood onset. N Engl J Med. (2007) 356(25):2603–13. doi: 10.1056/NEJMoa067597
14. Ghezzi A, Banwell B, Boyko A, Amato MP, Anlar B, Blinkenberg M, et al. Meeting review: the management of multiple sclerosis in children: a European view. Mult Scler. (2010) 16(10):1258–67. doi: 10.1177/1352458510375568
15. Tenembaum SN, Banwell B, Pohl D, Krupp LB, Boyko A, Meinel M, et al. Subcutaneous interferon Beta-1a in pediatric multiple sclerosis: a retrospective study. J Child Neurol. (2013) 28(7):849–56. doi: 10.1177/0883073813488828
16. Makhani N, Schreiner T. Oral dimethyl fumarate in children with multiple sclerosis: a dual-center study. Pediatr Neurol. (2016) 57:101–4. doi: 10.1016/j.pediatrneurol.2016.01.010
17. Ghezzi A, Pozzilli C, Grimaldi LME, Brescia Morra V, Bortolon F, Capra R, et al. Safety and efficacy of natalizumab in children with multiple sclerosis. Neurology. (2010) 75(10):912–7. doi: 10.1212/WNL.0b013e3181f11daf
18. Chitnis T, Arnold DL, Banwell B, Brück W, Ghezzi A, Giovannoni G, et al. Trial of fingolimod versus interferon Beta-1a in pediatric multiple sclerosis. N Engl J Med. (2018) 379(11):1017–27. doi: 10.1056/NEJMoa1800149
19. Vartzelis G, Maritsi D, Nikolaidou M, Garouf A, Kilidireas C. Rituximab as rescue therapy for aggressive pediatric multiple sclerosis. Case Rep Pediatr. (2019) 2019:1–3. doi: 10.1155/2019/8731613
20. McKeon A, Lennon VA, Lotze T, Tenenbaum S, Ness JM, Rensel M, et al. CNS aquaporin-4 autoimmunity in children. Neurology. (2008) 71(2):93–100. doi: 10.1212/01.wnl.0000314832.24682.c6
21. Chitnis T, Ness J, Krupp L, Waubant E, Hunt T, Olsen CS, et al. Clinical features of neuromyelitis optica in children: US network of pediatric MS centers report. Neurology. (2016) 86(3):245–52. doi: 10.1212/WNL.0000000000002283
22. Popescu BFG, Lennon VA, Parisi JE, Howe CL, Weigand SD, Cabrera-Gómez JA, et al. Neuromyelitis optica unique area postrema lesions: nausea, vomiting, and pathogenic implications. Neurology. (2011) 76(14):1229–37. doi: 10.1212/WNL.0b013e318214332c
23. Cacciaguerra L, Meani A, Mesaros S, Radaelli M, Palace J, Dujmovic-Basuroski I, et al. Brain and cord imaging features in neuromyelitis optica spectrum disorders: MRI and NMOSD diagnosis. Ann Neurol. (2019) 85(3):371–84. doi: 10.1002/ana.25411
24. Jarius S, Pellkofer H, Siebert N, Korporal-Kuhnke M, Hümmert MV, Ringelstein M. Cerebrospinal fluid findings in patients with myelin oligodendrocyte glycoprotein (MOG) antibodies. Part 1: results from 163 lumbar punctures in 100 adult patients. J Neuroinflammation. (2020) 17(1):261. doi: 10.1186/s12974-020-01824-2
25. Waters P, Jarius S, Littleton E, Leite MI, Jacob S, Gray B, et al. Aquaporin-4 antibodies in neuromyelitis optica and longitudinally extensive transverse myelitis. Arch Neurol. (2008) 65(7):913–9. doi: 10.1001/archneur.65.7.913
26. de Seze J. MOG-antibody neuromyelitis optica spectrum disorder: is it a separate disease? Brain. (2017) 140(12):3072–5. doi: 10.1093/brain/awx292
27. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. (2015) 85(2):177–89. doi: 10.1212/WNL.0000000000001729
28. Tenembaum S, Yeh EA, Guthy-Jackson Foundation International Clinical Consortium (GJCF-ICC). Pediatric NMOSD: a review and position statement on approach to work-up and diagnosis. Front Pediatr. (2020) 8:339. doi: 10.3389/fped.2020.00339
29. Koné-Paut I, Shahram F, Darce-Bello M, Cantarini L, Cimaz R, Gattorno M, et al. Consensus classification criteria for paediatric Behçet’s disease from a prospective observational cohort: PEDBD. Ann Rheum Dis. (2016) 75(6):958–64. doi: 10.1136/annrheumdis-2015-208491
30. Gallizzi R, Pidone C, Cantarini L, Finetti M, Cattalini M, Filocamo G, et al. A national cohort study on pediatric Behçet’s disease: cross-sectional data from an Italian registry. Pediatr Rheumatol Online J. (2017) 15(1):84. doi: 10.1186/s12969-017-0213-x
31. Hu YC, Chiang BL, Yang YH. Clinical manifestations and management of pediatric Behçet’s disease. Clin Rev Allergy Immunol. (2021) 61(2):171–80. doi: 10.1007/s12016-020-08809-2
32. Batu ED, Sonmez HE, Sozeri B, Butbul Aviel Y, Bilginer Y, Ozen S. The performance of different classification criteria in paediatric Behcet’s disease. Clin Exp Rheumatol. (2017) 35(Suppl 108):119–23.28406761
33. Koné-Paut I. Behçet’s disease in children: an overview. Pediatr Rheumatol Online J. (2016) 14:10. doi: 10.1186/s12969-016-0070-z
34. Saltik S, Saip S, Kocer N, Siva A, Yalçinkaya C. MRI findings in pediatric neuro-Behçet’s disease. Neuropediatrics. (2004) 35(3):190–3. doi: 10.1055/s-2004-815826
35. Moraleda-Cibrián M, Aracil-Martínez MA, Ercilla-González G, Ros-Viladoms J, Cusí-Sánchez V, González-Pascual E, et al. Neurobehçet de presentación precoz [Early onset Neurobehçet’s disease]. Rev Neurol. (2006) 42(3):150–2. doi: 10.33588/rn.4203.2005139
36. Hamzaoui K, Borhani-Haghighi A, Kaabachi W, Hamzaoui A. Increased interleukin 33 in patients with neuro-Behcet’s disease: correlation with MCP-1 and IP-10 chemokines. Cell Mol Immunol. (2014) 11(6):613–6. doi: 10.1038/cmi.2014.31
37. Uluduz D, Kürtüncü M, Yapıcı Z, Seyahi E, Kasapçopur Ö, Özdoğan H, et al. Clinical characteristics of pediatric-onset neuro-Behçet disease. Neurology. (2011) 77(21):1900–5. doi: 10.1212/WNL.0b013e318238edeb
38. Toker RT, Bodur M, Bican Demir A, Okan MS. Neuro-Behçet is a rare disease but should be considered in all kinds of neurological findings, even in childhood. Clin Exp Rheumatol. (2022) 40(8):1588–92. doi: 10.1016/B978-0-7020-4088-7.00110-3
39. Yavuz P, Solmaz I, Kaya UA, Akgoz A, Oguz KK, Aytac S, et al. Claude syndrome in childhood associated with probable neuro-Behcet disease. Neuropediatrics. (2023) 54(1):82–7. doi: 10.1055/s-0042-1759794
40. Metreau-Vastel J, Mikaeloff Y, Tardieu M, Koné-Paut I, Tran TA. Neurological involvement in paediatric Behçet’s disease. Neuropediatrics. (2010) 41(5):228–34. doi: 10.1055/s-0030-1269909
41. Koné-Paut I, Chabrol B, Riss JM, Mancini J, Raybaud C, Garnier JM. Neurologic onset of Behçet’s disease: a diagnostic enigma in childhood. J Child Neurol. (1997) 12:237–41. doi: 10.1177/088307389701200402
42. Ekici Tekin Z, Çelikel E, Aydin F, Kurt T, Sezer M, Tekgöz N, et al. Juvenile Behçet’s disease: a tertiary center experience. Clin Rheumatol. (2022) 41(1):187–94. doi: 10.1007/s10067-021-05896-0
43. Aviel YB, Batu ED, Sözeri B, Aktay Ayaz N, Baba L, Amarilyo G, et al. Characteristics of pediatric Behçet’s disease in Turkey and Israel: a cross-sectional cohort comparison. Semin Arthritis Rheum. (2020) 50(3):515–20. doi: 10.1016/j.semarthrit.2020.01.013
44. Shahram F, Nadji A, Akhlaghi M, Faezi ST, Chams-Davatchi C, Shams H, et al. Paediatric Behcet’s disease in Iran: report of 204 cases. Clin Exp Rheumatol. (2018) 36(Suppl 115):135–40.29998839
45. Nanthapisal S, Klein NJ, Ambrose N, Eleftheriou D, Brogan PA. Paediatric Behcet’s disease: a UK tertiary centre experience. Clin Rheumatol. (2016) 35:2509–16. doi: 10.1007/s10067-016-3187-z
46. Atmaca L, Boyvat A, Yalcindag FN, Atmaca-Sonmez P, Gurler A. Behcet disease in children. Ocul Immunol Inflamm. (2011) 19(2):103–7. doi: 10.3109/09273948.2011.555592
Keywords: Behcet’s disease, magnetic resonance imaging, children, early onset, acquired demyelinating syndromes (ADSs)
Citation: Pozzato M, Dilena R, Rogani G, Beretta G, Torreggiani S, Lanni S, Tozzo A, Andreetta F, Cavalcante P, Triulzi F, Martinelli Boneschi F, Minoia F and Filocamo G (2023) Can early-onset acquired demyelinating syndrome (ADS) hide pediatric Behcet's disease? A case report. Front. Pediatr. 11:1175584. doi: 10.3389/fped.2023.1175584
Received: 27 February 2023; Accepted: 30 May 2023;
Published: 23 June 2023.
Edited by:
Erkan Demirkaya, Western University, CanadaReviewed by:
Alessandro Capuano, Azienda Sanitaria Locale di Viterbo, ItalyFarhad Shahram, Tehran University of Medical Sciences, Iran
© 2023 Pozzato, Dilena, Rogani, Beretta, Torreggiani, Lanni, Tozzo, Andreetta, Cavalcante, Triulzi, Martinelli Boneschi, Minoia and Filocamo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giovanni Filocamo Z2lvdmFubmkuZmlsb2NhbW9AcG9saWNsaW5pY28ubWkuaXQ=
†These authors have contributed equally to this work