 Dan Zhao
Dan Zhao Ming Liu†
Ming Liu† Fang Fang
Fang Fang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 09 August 2023
Sec. Genetics of Common and Rare Diseases
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1173787
This article is part of the Research Topic Genetics of Inborn Errors of Metabolism View all 30 articles
Thiamine pyrophosphokinase (TPK) deficiency, is a rare autosomal recessive disorder of congenital metabolic dysfunction caused by variants in the TPK1 gene. TPK1 variants can lead to thiamine metabolic pathway obstacles, and its clinical manifestations are highly variable. We describe two cases of TPK deficiency with completely different phenotypes and different therapeutic effects, and 26 cases of previously reported were retrospectively reviewed to improve our understanding of the clinical and genetic features of the disease. Patients with TPK deficiency present with ataxia, dysarthria, dystonia, disturbance of consciousness, seizures, and other nervous system dysfunction. Different gene variant sites may lead to different clinical features and therapeutic effects. Gene analysis is important for the diagnosis of TPK deficiency caused by TPK1 variants, and thiamine supplementation has been the mainstay of treatment for TPK deficiency to date.
Thiamine (or vitamin B1) and its phosphorylate derivatives, thiamine monophosphate (TMP), thiamine pyrophosphate diphosphate (TPP), and thiamine triphosphate (TTP), are involved in energy metabolism and synthesis of nucleic acids, antioxidants, lipids, and neurotransmitters (1, 2). Different derivatives and metabolic pathways of thiamine are shown in Figure 1.
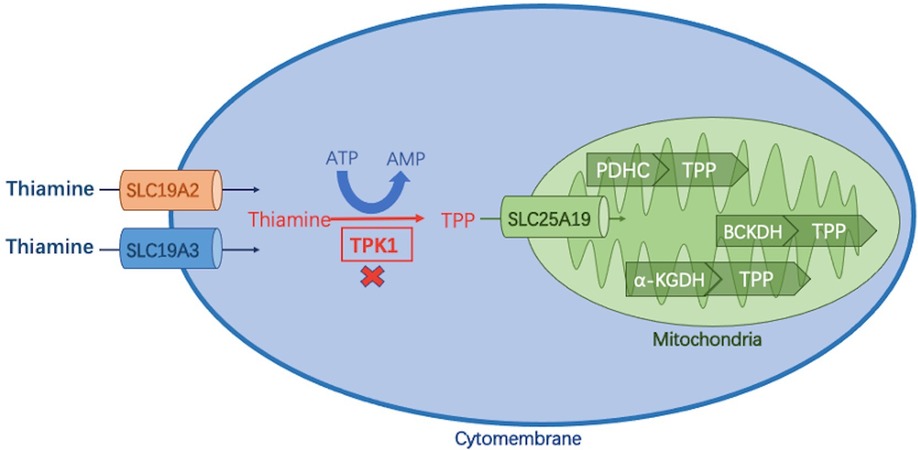
Figure 1. Thiamine metabolic pathway. Thiamine is absorbed in the small intestine and enters cells via two transporters (encoded by SLC19A2/SLC19A3), where it is phosphorylated to thiamine pyrophosphate (TPP) by thiamine pyrophosphate kinase 1 (TPK1). TPP enters the mitochondria via the SLC25A19-encoded carrier and acts as a cofactor of three different dehydrogenases to complete the energy metabolic pathway: (1) pyruvate dehydrogenase complex (PDHC); (2) branched α-ketoate dehydrogenase (BCKDH); α -ketoglutarate dehydrogenase (α-KGDH).
Four genes, SLC19A3 (OMIM#606152), SLC25A19 (OMIM#606521), SLC19A2 (OMIM # 603941), and TPK1 (OMIM#614458), are associated with thiamine metabolic disorders. Three of the genes SLC19A3, TPK1, and SLC25A19, are associated with acute encephalopathy, basal ganglia disease, and lactic acid accumulation caused by brain energy failure (3). Thiamine Metabolism Syndrome 5 (OMIM#614458) caused by TPK1 gene variants is a congenital thiamine metabolic disorder with a highly variable phenotype. The human TPK1 gene (hTPK1) is located on 7q35 with a full-length cDNA of 2,439 bp and encodes a protein composed of 243 amino acids. It contains nine exons and encodes proteins starting from the second exon. Thiamine pyrophosphate kinase 1 encoded by the TPK1 gene plays an important role as a thiamine activator in cells. TPK protein functions as a dimer, containing only one domain, and further enters mitochondria to play a role in oxidative phosphorylation of pyro phosphorylating thiamine into thiamine pyrophosphate (TPP) (4).TPP is an active form of thiamine and a key cofactor of several enzymes that play major roles in energy metabolism (5, 6). Compared with patients with SLC19A3 and SLC25A19, patients with TPK1 variants presented with symptoms earlier, responded differently to thiamine supplementation, and developed a more severe phenotype with higher rates of morbidity and mortality (7). According to previous reports, not all patients with TPK1 defects responded to thiamine therapy. The two previously unreported TPK1 variants in this study elicited distinct responses to thiamine supplementation, which may be related to their age at onset, phenotype, and early start of high doses of parenteral thiamine treatment. Here, we describe two cases of TPK deficiency with completely different phenotypes and different therapeutic effects. Additionally, through literature search and analysis, 26 cases of previously reported patients were retrospectively analyzed to improve our understanding of the clinical and genetic features of the disease.
In this study, two patients with TPK1 deficiency were recruited from the Department of Neurology of Beijing Children's Hospital. Both of them had significant neurological symptoms and were diagnosed by whole exon sequencing (WES).
The keywords, “TPK1”, “TPK deficiency”, “thiamine pyrophosphatase deficiency”, and “Thiamine pyrophosphatase deficiency”, were used as search terms in Embase, PubMed database, CNKI database, and Wanfang Database for literature review. A total of 209 articles were retrieved after removing duplicate items, which were retrieved from the database built up to February 2022. After excluding articles with incomplete cases, articles without genetic diagnoses, and irrelevant articles, 17 articles comprising 26 cases (5–20) were included (screening process is shown in Figure 2). Including the two cases reported in this study, we have shown the phenotype, genotype, clinical information, and treatment of 28 patients in the Supplementary Material.
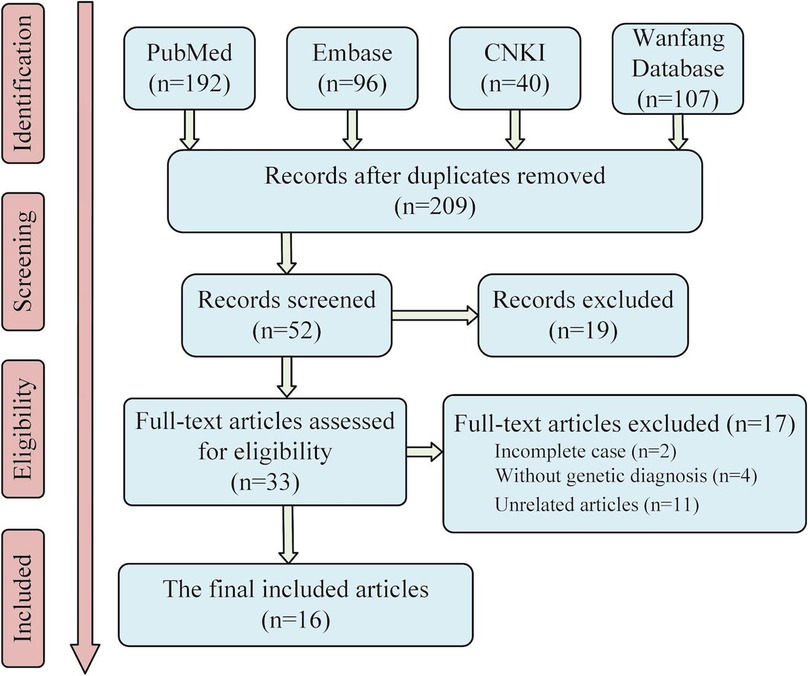
Figure 2. Literature screening flow chart. Sixteen articles were finally included in the analysis. Considering the two cases reported in this paper, twenty-eight patients with TPK1 gene defect were included in this study.
All patients reported in the literatures were classified into Leigh group and non-Leigh group based on whether they had bilateral symmetric basal ganglia or cerebellar lesions on brain MRI. All patients treated with thiamine were divided into effective group (obvious improvement and certain improvement) and ineffective group (no improvement and death) according to their therapeutic efficacy.
Continuous variables were summarized with standard descriptive measures, including means, standard deviation, median, and interquartile ranges, whereas categorical variables were summarized using frequencies and percentages. For continuous variables, differences among comparison groups were estimated using the student t-test for normally distributed variables and the Wilcoxon rank-sum test for variables that were not normally distributed. Differences in categorical variables were assessed using the Chi-square test or the Fisher's Exact test. A p-value of <0.05 was considered significant. All statistical analyses were performed using IBM SPSS Statistics 22 software (IBM Corp., Armonk, NY).
A 1-year and 8 months old boy first presented with unsteady gait and dysarthria of a week's duration. Before the onset, the patient had a fever of 39°C, rashes, and a convulsive seizure. Afterwards, unsteady gait and dysarthria, appeared and progressively worsened. The patient had no other infectious symptoms, such as cough, vomiting, and diarrhea. He had no hearing loss, intellectual disability, or other symptoms. He was the second child of a nonconsanguineous family and uneventfully delivered naturally at full term. His birth weight was 3 kg, and he had a suspicious history of intrauterine hypoxia at birth. Developmental milestones were normal before onset.
Physical examination showed unsteady walking and sitting, wide-based gait, dystonia, and dysarthria. Other cranial nerve examinations were unremarkable. Bilateral tendon reflexes were symmetrical; muscle strength and tone were normal; and pyramidal signs and meningeal irritation signs were negative.
Blood lactic acid, blood ammonia, and homocysteine levels were normal. Urine screening showed mild keto-dicarboxylic acid urine with slightly higher levels of several organic acids. Blood screening showed no obvious abnormalities. Brain MRI showed symmetrical patchy long T2 signals in the cerebellar dentate nucleus, bilateral basal ganglia, and thalamus (Figure 3A). Gesell Developmental Assessment was performed, and cognitive area and personal-social area were normal. The patient had a mild developmental delay in the adaptation area, gross motor area, and fine motor area. A mitochondrial genetic test revealed a negative finding.
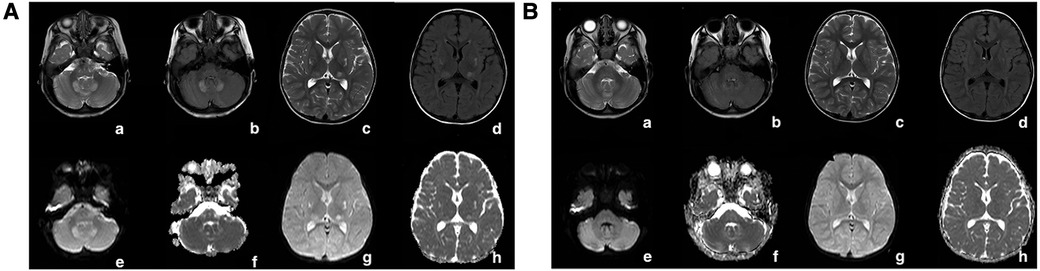
Figure 3. (A) In patient 1, at the onset of the disease (1 year and 8 months old), the cerebral MRI showed symmetrical patchy long T2 signals in the cerebellar dentate nucleus, bilateral basal ganglia, and thalamus. (B) In patient 1, the signal of long T2 in the brain improved gradually after vitamin B1 treatment for 1 year. The long patchy T2 signals were significantly attenuated on T2WI (a,e), T2-FLAIR (b,c), and DWI (c,g) sequences.
Leigh Syndrome was diagnosed and treated with cocktail therapy (thiamine 100 mg/day, riboflavin 100 mg/day, vitamin C 50 mg/day, vitamin E 100 mg/day, and coenzyme Q10 90 mg/day). After 2 months of treatment, his symptoms improved and he was followed up in the outpatient department for a long period. After 1 year of treatment, multiple abnormal signals previously observed in the brain MRI had reduced (Figure 3B). At the age of 4 years, symptoms recurred with ataxia, dysarthria, and inability to walk unaided after discontinuation of the drug. The symptoms gradually resolved after repeating the drug regimen (thiamine 100–200 mg/day oral treatment). Whole exon sequencing (WES) was performed after the parents agreed and signed informed consent. Compound heterozygous variants of TPK1 in the patient (NM_022445.3), c.513delG (p.Arg171Ser fs*3) and c.382C>T (p.Leu128Phe), were identified (Figures 4A,B). The WES results supported the diagnosis of TPK1-associated Leigh syndrome. Oral treatment with a large dose of thiamine (10 mg/kg·day) was continued with the discontinuation of other medications.
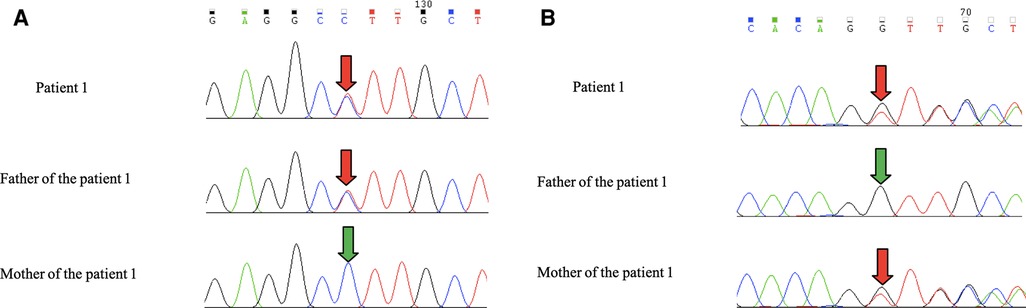
Figure 4. (A,B) Patient 1 had the c.513delG + c.382C>T complex heterozygous variant. The father carried the TPK1 gene c.382C>T heterozygous variant, and the mother carried the TPK1 gene c. 513delG heterozygous variant.
The patient was followed up regularly annually. In the past 2 years, due to the COVID-19 pandemic, in-person follow-up was replaced by online video consultations. At the age of 6 years and 7 months, he discontinued against medical advice for 1 month, leading to a recurrence of the symptoms of walking instability and dysarthria. The symptoms disappeared after 2–3 days of remedication (200 mg/day). At the last follow-up, the patient was 9 years and 2 months old. He took thiamine regularly and had a normal life without ataxia and other symptoms.
An 8 months old male infant first reported to the hospital with developmental delay after birth and convulsions from the age of 1 month. The patient had no definite infectious history before the onset. At the age of 1 month old, seizures were tonic seizures, which lasted about 1–2 min, and occurred 4–5 times per month. At the age of 4 months, seizures became spasms, which were 30–40 clusters per event, and occurred once or twice per week. Developmental milestones were delayed after birth. He could not recognize and move towards the direction of sound and objects, raise his head, turn over, or sit. He was the second child of his parents, had a healthy sister, and his mother had an abortion. Family and birth histories were normal.
Physical examination revealed a microcephaly, inability to chase sound and objects, occasional nystagmus, and dystonia with hands clenched and thumbs adducted, presence of symmetrical bilateral patellar tendon reflexes, negative meningeal irritation sign, limb muscle power of 4, and hypertonia.
Metabolic analysis of blood showed a lactate level of 3.8 mmol/L and high level of pyruvate (0.2 mmol/L). Organic acid in urine analysis showed significantly increased levels of urine 2-ketoglutarate and several organic acids. The activity of mitochondrial respiratory chain enzyme complexes was normal. At the age of 5 months, a muscle biopsy was performed and muscle pathology showed slight pathological changes. These changes included presence of hypertrophic, atrophic, and necrotic myofibers; increased poisoning of fat droplets in myofibers, and increased number of MHC-1-positive muscle fibers. The electroencephalogram showed an atypical hypsarrhythmia, and tonic seizures and clusters of spasms were observed. Brain MRI showed abnormal signals in the head of the caudate nuclei, lenticular nuclei, and dentate nuclei of the cerebellar hemisphere (Figure 5). WES was performed after the parents agreed and signed informed consent. Compound heterozygous variants of TPK1, (NM_022445.3) c. 263G>A (p.Cys88Tyr) and c.465_467delAAT (p. Leu156del), were identified.
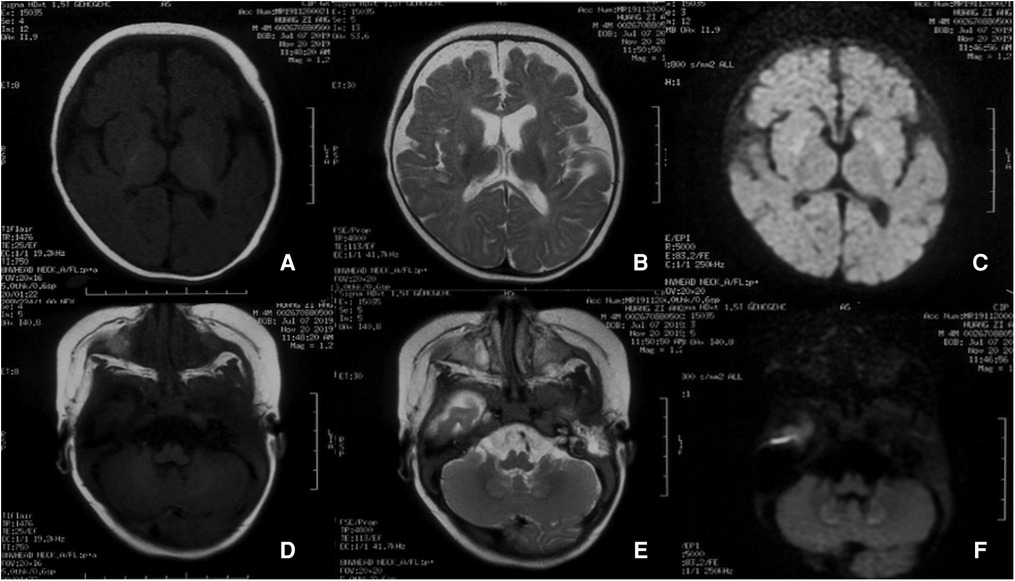
Figure 5. Brain MRI of case 2 at the beginning of the disease. Presence of roughly symmetrical abnormal signal foci in the head of the caudate nuclei, lenticular nuclei, and dentate nuclei of the cerebellar hemisphere.
Several antiepileptic drugs, such as topiramate, levetiracetam, and vigabatrin, were used to control the seizures but were less effective. The patient underwent concurrent cocktail therapy, including oral administration of thiamine (15–20 mg/kg·day), L-carnitine, biotin, coenzyme Q10, vitamin C, and vitamin E. At the age of 10 months, the patient underwent another brain MRI, which showed that the initial lesion had disappeared, the brain had atrophied, and the ventricles had enlarged. At the age of 1 year, although receiving daily doses of thiamine 100 mg intramuscularly and 100 mg orally, the patient could not chase sound and objects and raised his head unstably. More than 10 episodes of seizures were observed per day.
At the age of 1 year and 1-month-old, the patient was fed with a ketogenic diet (ratio 3:1), which reduced frequency of seizures. However, the treatment was discontinued at the age of 1 year and 7 months due to intolerance to the ketogenic diet. The patient died of encephalopathy and respiratory failure at the age of 2 years and 3 months.
The 28 patients included twelve (42.86%) males, fourteen (50%) females, and two (7.14%) unknown genders. Among them, nine (32.14%) patients had consanguineous parents. In terms of country and area distribution, thirteen (46.43%) patients were from China, five (14.29%) patients were from Europe (two cases from Germany, one case from Finland, and one case from Switzerland), three (10.71%) patients from the United States, two (7.14%) patients from Iraq, and one (3.57%) patient from India. The nationalities of five (17.86%) patients were unknown (Figures 6A,B).
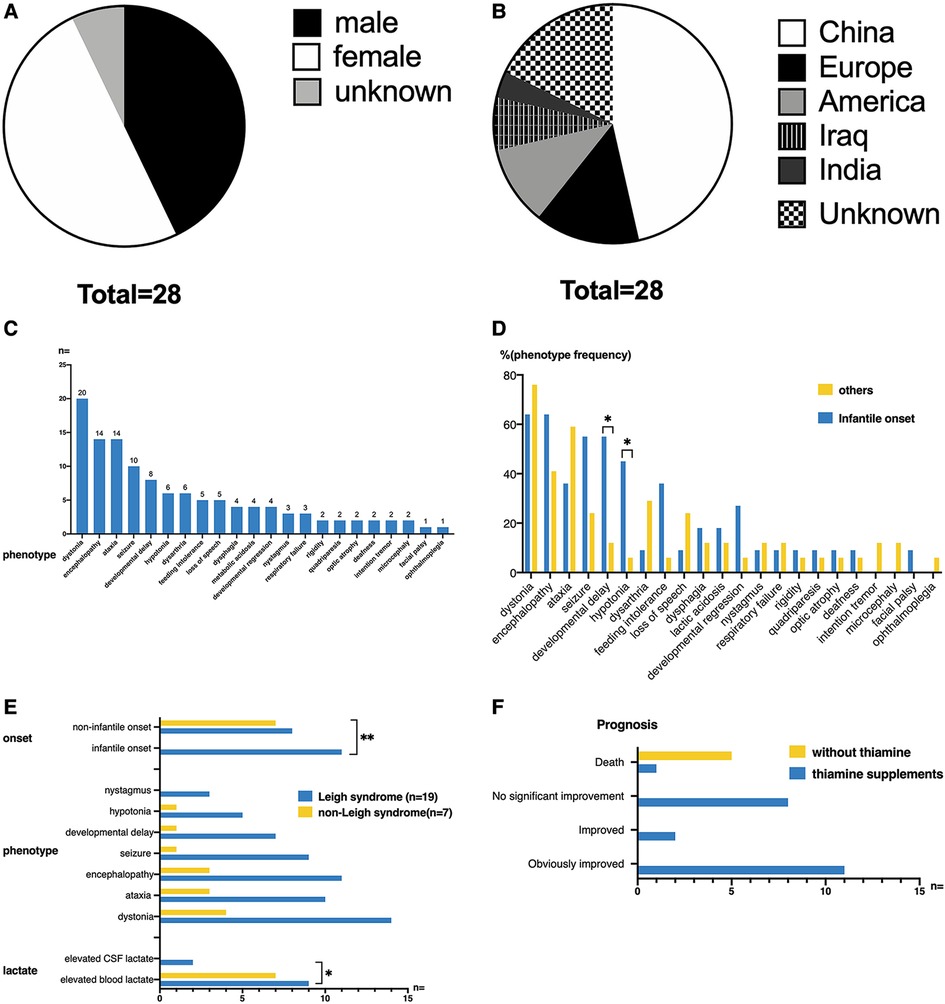
Figure 6. (A,B) Gender distribution and ethnic origin of all published cases of TPK1 gene defect. (C) The clinical phenotypes of twenty-eight patients. The most common clinical manifestations included dystonia, ataxia, encephalopathy, seizure, and developmental delay. The most common signs included hypotonia and nystagmus. (D) Phenotypic differences between infantile-onset and non-infantile-onset patients. Among all phenotypes, developmental delay and hypotonia were more common in patients with infantile-onset (Fisher's Exact Test, p < 0.05). (E) Differences in clinical features between patients with Leigh syndrome and non-Leigh syndrome. The onset age of patients with Leigh syndrome was more distributed in the infantile onset group (Fisher's Exact Test, p = 0.01). Elevated blood lactate level was more common in the non-Leigh syndrome group (Fisher's Exact Test, p < 0.05). (F) Thiamine supplementation in patients with TPK1 gene variants and related outcomes. Among the twenty-eight patients, twenty-two patients received thiamine supplementation (20–750 mg/day), eleven patients had significant improvement in clinical symptoms, two patients had partial improvement, and nine patients had no significant improvement [one of these nine patients (case 2 of this study) died].
For these patients, the onset of disease was mostly during infancy (11 cases, 39.29%), early childhood (11 cases, 39.29%), early school age (5 cases, 17.85%), and unknown (1 case, 3.57%). Twenty (71.43%) cases were preceded by an infection or trauma. The clinical phenotypes of the 28 patients are shown in Figure 6C. The most common clinical manifestations were dystonia (20/28, 71.43%), ataxia (14/28, 50%), encephalopathy (14/28, 50%), seizure (10/28, 35.71%), and developmental delay (8/28, 28.57%). The most common signs included hypotonia (6/28, 21.43%) and nystagmus (3/28, 10.71%). Other symptoms and signs observed in few patients were rigidity (2/28, 7.14%), quadriparesis (2/28, 7.14%), optic atrophy (2/28, 7.14%), deafness (2/28, 7.14%), intention tremor (2/28, 7.14%), microcephaly (2/28, 7.14%), facial palsy (1/28, 3.57%), and ophthalmoplegia (1/28, 3.57%).
According to age of onset, patients were divided into an infantile-onset group and non-infantile onset group (as shown in Figure 6D). Among all phenotypes, developmental delay and hypotonia were more common in patients with infantile onset, and the differences were statistically significant (p < 0.05).
Blood lactic acid tests performed in 26 cases revealed elevations in sixteen (16/26, 61.53%) cases (reference range: 0.5–1.7 mmol/L), and lactic acidosis during the course of the disease in two cases. Only two (2/14, 14.29%) patients had elevated levels of cerebrospinal fluid (reference range: 1.0–2.8 mmol/L). The levels of thiamine pyrophosphate (TPP) in blood and muscle were measured in some patients. Muscular concentrations of TPP were decreased in all five patients (5/5, 100%), and TPP concentrations in blood were decreased in eight patients (8/12, 66.67%). Organic acid in urine analysis was performed in nineteen patients; eleven patients (11/19, 57.89%) showed elevated urine 2-KGA levels, and six patients (6/19, 31.58%) had normal levels of urine 2-KGA. Pyruvate levels were measured in eight patients, that were elevated in six patients (75%). The skin/muscle respiratory chain complex activity was measured in nine patients (P2, P3, P5–P10, P16), which revealed slightly decreased (3/9, 33.33%) or normal activity (6/9, 66.67%). We observed a slight decrease in complex V enzyme activity in P2 muscles; complex I, complex II, complex III, and complex I + III in P3 muscles; and complex III in P5 muscles.
Brain MRIs were performed in 26 patients at the acute stage. Five patients had normal cerebral MRI findings, whereas twenty-one patients (80.77%) had abnormal brain MRI findings, which mainly involved the bilateral basal ganglia (50%), thalamus (23.08%), and especially cerebellum (50%). In few cases, the brainstem, corpus callosum, and spinal cord were involved. A diagnosis of Leigh syndrome was made in nineteen cases (73.08%). Brain MRI findings in seven cases (26.92%) showed that original lesions improved or disappeared after thiamine treatment.
According to the presentation of cerebral MRI, 26 children with acute state MRIs were divided into the Leigh syndrome group (19/26, 73.08%) and the non-Leigh syndrome group (7/26, 26.92%). As shown in Figure 6E, the onset age of patients with Leigh syndrome was highly distributed during infancy (p = 0.01). No significant differences in symptoms and signs were observed between the Leigh syndrome group and the non-Leigh syndrome group. Elevated blood lactate was more common in the non-Leigh syndrome group (p < 0.05).
As shown in Figure 6F, twenty-two patients received thiamine supplementation (20–750 mg/day), five patients did not receive thiamine supplementation, and one patient did not mention treatment and prognosis. Among patients who received thiamine supplementation, eleven (11/22, 50%) patients had significant improvement in clinical symptoms, two (2/22, 9.09%) patients had partial improvement, nine (9/22, 4.91%) patients had no significant improvement, in which one patient (1/22, 4.55%) died. All five patients who did not receive thiamine supplementation died. The survival rate was higher in the thiamine-treated group (21/22, 95.5%) than in the thiamine-untreated group (0/6, 0.0%) (p < 0.01). Among the six dead patients, two patients had unknown causes, one patient died of viral infection at 3.5 years old, and three other patients with clinically severe recurrent encephalopathy and refractory epilepsy died of metabolic crisis, respiratory failure, and multi-organ failures. The mean age at death was 4.72 years, and the median age was 2.96 years (0.5–11 years).
Twenty-two patients who received thiamine treatment were divided into the effective group (thirteen cases with significant clinical improvement or partial improvement) and the ineffective group (nine cases with clinical failure or death) (Figure 7). No difference in age of onset was observed between the two groups (1.64 ± 1.18 vs. 1.94 ± 1.83, t-test: t = –0.470, p > 0.1). The maximum reported age of survival was 40 years old. In terms of clinical symptoms, ataxia onset was more common in the effective group (84.62% vs. 11.11%, p < 0.005), whereas seizures were more common in the ineffective group (4/9, 44.44%). Although the basal ganglia involvement rate in the ineffective group (7/9, 77.78%) was higher than that in the effective group (6/13, 46.15%), no difference was observed (p > 0.05). The rate of increase in blood lactic acid level in the two groups was more than 60%; however, no significant difference was observed. Additionally, no significant difference in the proportion of patients with Leigh syndrome was observed between the effective group and ineffective group.
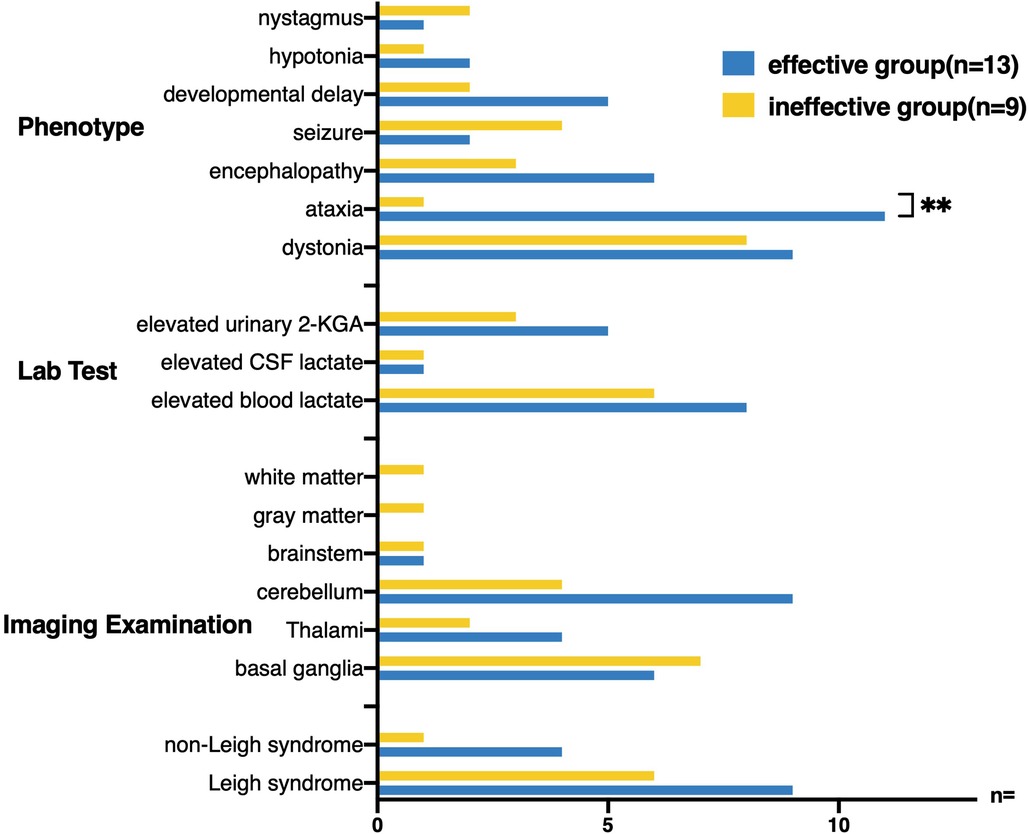
Figure 7. Twenty-two patients receiving thiamine treatment were divided into effective treatment group and ineffective treatment group, and the clinical characteristics of the two groups were compared. Ataxia onset was more common in the effective treatment group (84.62% VS 11.11%, Fisher’s exact test, p < 0.005), whereas seizures were more common in the ineffective treatment group (4/9, 44.44%). The cerebellum (9/13, 69.23%) was more likely to be involved in the brain MRI in the effective treatment group.
The distribution of all discovered TPK1 variants of the gene, including the four variants in this study, is shown in Figure 8. Twenty-two gene variants of the TPK1 gene were found, and most of them were located in exon 7, followed by exon 3 and exon 2. Among the 22 gene variants, 15 missense variants, three splicing variants, three frameshift variants, and one fragment deletion variant were identified.
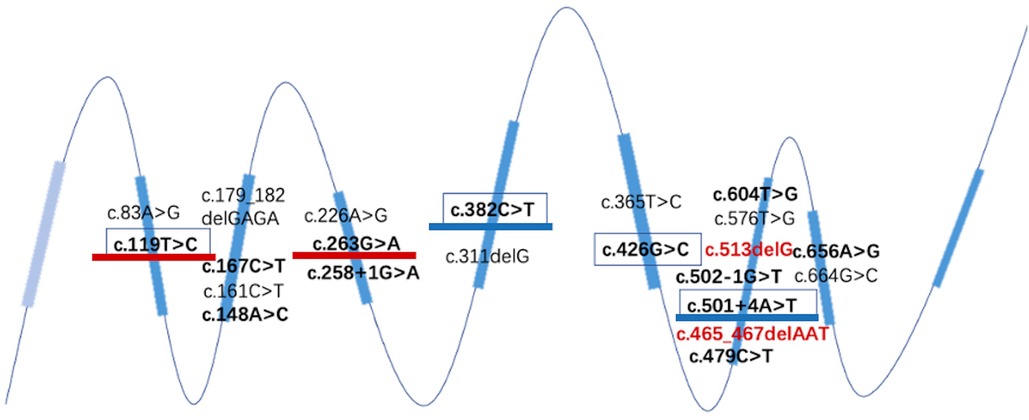
Figure 8. Structure of all reported TPK1 gene variant loci. Red font represents the newly discovered variants in this study, all located in the seventh exon. The variants present in ≥3 cases have been highlighted. Effective therapeutic variants have been underlined in blue. Therapy-effective variants are underlined in blue. The sites of variants that did not respond to treatment are underlined in red.
Of the twenty-eight patients, three had frameshift mutations confirmed by WES, and two of them received thiamine therapy, all of which were effective. There were twenty-five cases of non-frameshift mutation, of which sixteen were treated with thiamine and nine were effective (p > 0.05).
Among the twelve patients who underwent thiamine treatment, four patients had the c. 382C>T (P. Lys128Phe) variant and two patients had the C. 501 + 4A > T (p. Val119_Pro167del) variants (underlined in blue in Figure 6). Among the eight patients who did not respond to treatment, two and three patients had c.263G>A (p. Cys88Tyr) and C. 119T>C (p. lys40Pro), respectively (underlined in red in Figure 8). Patients who died had exon 3, 4, 6, or 7; and no aggregation of death cases was found in a specific exon or variant site.
Herein, we report two cases of THMD5 with different clinical manifestations and different responses to thiamine treatment of TPK1 variants. One patient presented with ataxia, and thiamine replacement therapy could achieve significant clinical efficacy. The other patient had refractory epilepsy, and thiamine combined with antiepileptic therapy had a poor prognosis.
Up to now, twenty-six patients from thirteen families, five of which are consanguineous, had been reported before the publication of this manuscript. The mean onset age of thiamine pyrophosphokinase deficiency caused by TPK1 variants is 1.85 years. The cases of TPK1 variants are most likely to occur in infants, but the mean time of diagnosis is approximately 5 years. Compared with other mitochondrial diseases, TPK1 variants has various clinical phenotypes without specific clinical characteristics. In addition to the main symptoms, such as ataxia, encephalopathy, and dystonia, it can be combined with feeding intolerance and developmental delay or regression. Children with infantile onset of the disease are more likely to have developmental problems, whereas others are more likely to show ataxia. No significant difference in the age of onset of epilepsy and epileptic encephalopathy were observed.
Elevated lactate levels in the blood and cerebrospinal fluid were generally considered to be auxiliary diagnostic methods. A total of 61.5% of patients with TPK deficiency had elevated blood lactate level and only 21.4% of patients had elevated cerebrospinal fluid lactate level. Therefore, elevated blood/cerebrospinal fluid lactate seems not to be a specific biomarker of TPK1 variants. In the patients who underwent the TPP concentration test, the blood/muscle TPP concentration decreased. Therefore, the decline in TPP concentration can be used as a specific biomarker for TPK defects, but it cannot be routinely tested in clinical practice due to the high level of requirements needed to establish for detection facilities. Organic acid in urine analysis revealed that urine 2-KGA level was elevated in more than half of the patients, which has certain suggestive significance for diagnosis. Since TPK defect affect pyruvate thiamine metabolism by leading to a lack of key enzyme cofactors, it can show a decrease in pyruvate level and PDC activity. However, the TPP does not affect the mitochondrial OXPHOS respiratory chain protein expression and activity, resulting in normal respiratory chain complex enzymes (10). This finding is consistent with findings from previously reported cases with high blood pyruvate level but normal respiratory chain enzyme activity.
A noteworthy finding was the presence of cerebellar involvement as the main imaging feature of the presence of TPK1 variants. Based on imaging characteristics, patients were grouped into Leigh syndrome group and non-Leigh syndrome group. Patients in the Leigh syndrome group had a younger onset age and less elevated level of lactate in blood than the non-Leigh syndrome group. These findings are not often mentioned in previous literature. Whether this phenomenon is related to the difference in specific gene variation sites needs further detailed study.
TPK deficiency due to TPK1 variants is a treatable disease. Some patients had mild clinical manifestations, especially those with ataxia. Thiamine supplementation immediately after early diagnosis or suspected diagnosis showed significant improvement or even normalization of clinical symptoms. However, failure to receive timely thiamine treatment can lead to irreversible brain damage and catastrophic consequences. In this study, death was recorded only among patients who were untreated. The only patient who received thiamine treatment but still had death as clinical outcome was the patient in case 2 in this study. This child had an early onset age and presented with severe epilepsy and encephalopathy clinically, but thiamine supplement therapy had no significant effect. Late treatment with a ketogenic diet was not well tolerated. TPK deficiency requires early recognition as one of the few treatable mitochondrial diseases. Suspected mitochondrial diseases, especially thiamine metabolic deficiencies, should be treated early with a thiamine cocktail in combination with other multivitamins. Thiamine should be dosed after genetic confirmation, but no standards for the recommended dose of thiamine are currently available. Most medical institutions use thiamine 100–400 mg/day (or 10 mg/kg/day). Larger long-term studies are needed to determine the appropriate dose of thiamine supplementation during and between onset. Additionally, Mayr et al. (8) proposed direct supplementation of TPP as the synthesis defect of TPP, which is a key component of the pathogenicity of TPK1 variant. However, the effectiveness and safety of TPP have yet to be confirmed.
In this study, two new variants of the TPK1 gene were reported; c. 513delG (p. Arg171SerFS *4) and c.465_467delAAT (p. Leu156del) were both frame-shift variants, which resulted in changes in the amino acid triad coding frame, thus affecting the structure and function of the coding protein. The ACMG ratings of the two variants were both potentially pathogenic. This site has never been reported by previous studies. Two cases of c. 382C>T (p. Leu128Phe) have been reported as a pair of twins who showed paroxysmal ataxia (c. 382C>T homozygous variant). L128F is a variant on a highly conserved residual, and L128 is located in mice's α/β domain of TPK crystal structure. It is related to the formation of TPK dimer and may destroy the function of the enzyme (21).
Few studies on the pathogenesis of TPK1 variant have been reported. Banka et al. (9) reported that the enzyme activities of S160l and D222H mutants were lower than that of wild-type TPK1, but Huang et al. (13) found that the enzyme activity of most mutants was significantly reduced by testing the enzyme activity of various TPK1 mutants, whereas the activities of S160l and W202G mutants were significantly higher than that of the wild-type. Furthermore, isothermal titration calorimetry was used to determine whether the variant affected the affinity of TPK1 to thiamine. The affinity of S160l to thiamine was significantly reduced, and no bond between the W202G mutant and thiamine was detected. The decrease in bonds of S160l and W202G to TPP, a thiamine structurally similar product, increased enzyme activity. Additionally, Huang et al. (13) measured the melting temperatures (Tm) of the wild-type and mutant TPK1 by differential scanning calmetry and found that the protein stabilities of four mutants (S160l, W202G, N219S, and D222H) were significantly lower than that of the wild-type. They suggest that the TPK1 variant can alter thiamine metabolism by altering TPK1 enzyme activity, reducing the affinity of TPK1 for thiamine, or reducing the stability of TPK1 protein (13). However, no cytological or animal studies have provided further evidence.
In the conclusion of a report on the prognosis of 22 thiamine-treated patients, no difference in onset age was observed between the effective treatment and ineffective treatment groups. More cases of ataxia was observed in the effective group, and brain MRI showed more cerebellar involvement. The variants, c. 382C>T (p. Lys128Phe) (17, 22) and c. 501 + 4A > T (P. Val119_Pro167del) (23, 24), were discovered more in the effective treatment group. c.263G > A (p. Cys88Tyr) (14) and C.119T > C (p. Lys40Pro) (8, 12) were more common in the ineffective treatment group. This finding is consistent with the finding of a difference in the therapeutic effect of the two cases in our study. Currently, few cases have been reported and variant sites are scattered, and no obvious correlation between genotype and clinical phenotype has been found. The specific mechanism needs to be further studied.
The clinical manifestations of THMD5 are highly variable. Therefore, the early diagnosis of TPK deficiency is a huge challenge. The clinical manifestations are acute onset encephalopathy or ataxia and unexplained seizures, especially with Leigh syndrome starting in infancy. Active and well-developed WES facilitates early molecular diagnosis. Meanwhile, for THMD5, a rare neurodegenerative disease, early supplementation with thiamine has a great effect on prognosis.
The original contributions presented in the study are included in the article and supplementary table, further inquiries can be directed to the corresponding authors.
Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
DZ, HJ, and CX collected the clinical data of patients. HJ, ML, ZL, and FF contributed to genetic analysis. DZ, ML, HJ, TS, CX, XD, RD, and HX provided help in the clinical evaluation of the patients. JW and ML performed routine follow-ups of the patients and drafted the manuscript. FF and ZL designed the study and contributed to English editing of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Nation Natural Science Foundation of China (82001791).
We thank the patients and their families who participated in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2023.1173787/full#supplementary-material
1. Tittmann K. Reaction mechanisms of thiamin diphosphate enzymes: redox reactions. FEBS J. (2009) 276(9):2454–68. doi: 10.1111/j.1742-4658.2009.06966.x
2. Sechi GP, Bardanzellu F, Pintus MC, Sechi MM, Marcialis MA, Fanos V. Thiamine as a possible neuroprotective strategy in neonatal hypoxic-ischemic encephalopathy. Antioxidants (Basel). (2021) 11(1):42–42. doi: 10.3390/antiox11010042
3. Marce-Grau A, Marti-Sanchez L, Baide-Mairena H, Ortigoza-Escobar JD, Perez-Duenas B. Genetic defects of thiamine transport and metabolism: a review of clinical phenotypes, genetics, and functional studies. J Inherit Metab Dis. (2019) 42(4):581–97. doi: 10.1002/jimd.12125
4. Zhao FGR, Goldman ID. Molecular cloning of human thiamin pyrophosphokinase. Biochim Biophys Acta. (2001) 1517(2):320–2. doi: 10.1016/S0167-4781(00)00264-5
5. Losa R, Sierra MI, Fernandez A, Blanco D, Buesa JM. Determination of thiamine and its phosphorylated forms in human plasma, erythrocytes and urine by HPLC and fluorescence detection: a preliminary study on cancer patients. J Pharm Biomed Anal. (2005) 37(5):1025–9. doi: 10.1016/j.jpba.2004.08.038
6. Sechi G, Serra A. Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet Neurol. (2007) 6(5):442–55. doi: 10.1016/S1474-4422(07)70104-7
7. Ortigoza-Escobar JD, Alfadhel M, Molero-Luis M, Darin N, Spiegel R, de Coo IF, et al. Thiamine deficiency in childhood with attention to genetic causes: survival and outcome predictors. Ann Neurol. (2017) 82(3):317–30. doi: 10.1002/ana.24998
8. Mayr JA, Freisinger P, Schlachter K, Rolinski B, Zimmermann FA, Scheffner T, et al. Thiamine pyrophosphokinase deficiency in encephalopathic children with defects in the pyruvate oxidation pathway. Am J Hum Genet. (2011) 89(6):806–12. doi: 10.1016/j.ajhg.2011.11.007
9. Banka S, de Goede C, Yue WW, Morris AA, von Bremen B, Chandler KE, et al. Expanding the clinical and molecular spectrum of thiamine pyrophosphokinase deficiency: a treatable neurological disorder caused by TPK1 mutations. Mol Genet Metab. (2014) 113(4):301–6. doi: 10.1016/j.ymgme.2014.09.010
10. Fraser JL, Vanderver A, Yang S, Chang T, Cramp L, Vezina G, et al. Thiamine pyrophosphokinase deficiency causes a leigh disease like phenotype in a sibling pair: identification through whole exome sequencing and management strategies. Mol Genet Metab Rep. (2014) 1:66–70. doi: 10.1016/j.ymgmr.2013.12.007
11. Invernizzi CP F, Chiapparini L, Moroni I, Nardocci N, Garavaglia B, Tonduti D. Thiamine-responsive disease due to mutation of TPK1: importance of avoiding misdiagnosis. Neurology. (2017) 9:870–971. doi: 10.1212/WNL.0000000000004270
12. Mahajan A, Sidiropoulos C. TPK1 Mutation induced childhood onset idiopathic generalized dystonia: report of a rare mutation and effect of deep brain stimulation. J Neurolog Sci. (2017) 376:42–3. doi: 10.1016/j.jns.2017.02.063
13. Huang W, Qin J, Liu D, Wang Y, Shen X, Yang N, et al. Reduced thiamine binding is a novel mechanism for TPK deficiency disorder. Mol Genet Genomics. (2019) 294(2):409–16. doi: 10.1007/s00438-018-1517-3
14. Zhu L, Wu R, Ye Z, Gu R, Wang Y, Hou Y, et al. Identification of two novel TPK1 gene mutations in a Chinese patient with thiamine pyrophosphokinase deficiency undergoing whole exome sequencing. J Pediatr Endocrinol Metab. (2019) 32(3):295–300. doi: 10.1515/jpem-2018-0363
15. Nyhan WL, McGowan K, Barshop BA. Thiamine phosphokinase deficiency and mutation in TPK1 presenting as biotin responsive basal ganglia disease. Clin Chim Acta. (2019) 499:13–5. doi: 10.1016/j.cca.2019.07.034
16. Bugiardini E, Pope S, Feichtinger RG, Poole OV, Pittman AM, Woodward CE, et al. Utility of whole blood thiamine pyrophosphate evaluation in TPK1-related diseases. J Clin Med. (2019) 8(7):991. doi: 10.3390/jcm8070991
17. Zhu B, Wu J, Chen G, Chen L, Yao Y. Whole exome sequencing identifies a novel mutation of TPK1 in a Chinese family with recurrent ataxia. J Mol Neurosci. (2020) 70(8):1237–43. doi: 10.1007/s12031-020-01568-x
18. Li D, Song J, Li X, Liu Y, Dong H, Kang L, et al. Eleven novel mutations and clinical characteristics in seven Chinese patients with thiamine metabolism dysfunction syndrome. Eur J Med Genet. (2020) 63(10):104003. doi: 10.1016/j.ejmg.2020.104003
19. Au LWC, Lee HHC, Sheng B, Chan KY, Yau EKC, Mak CM, et al. Movement disorders associated with thiamine pyrophosphokinase deficiency: intrafamilial variability in the phenotype. Clin Neurol Neurosurg. (2020) 199:106258. doi: 10.1016/j.clineuro.2020.106258
20. Jue W, Xiane C, Ainong M. Analysis of clinical characteristics and gene variation in a family with thiamine pyrokinase deficiency. Chinese J Genet. (2021) 38(2):141–4. doi: 10.3760/cma.j.cn511374-20191208-00627
21. Bacchelli C, Williams HJ. Opportunities and technical challenges in next-generation sequencing for diagnosis of rare pediatric diseases. Expert Rev Mol Diagn. (2016) 16(10):1073–82. doi: 10.1080/14737159.2016.1222906
22. Shaowei L, Lizhi Z, Hai Y, Xianrui C. Clinical characteristics and genetic analysis of a child with thiamine metabolic dysfunction syndrome type 5. Chinese J Med Genet. (2021) 38(9):873–6. doi: 10.3760/cma.j.cn511374-20200428-00310
23. Rusch CT, Wortmann SB, Kovacs-Nagy R, Grehten P, Haberle J, Latal B, et al. Thiamine pyrophosphokinase deficiency due to mutations in the TPK1 gene: a rare, treatable neurodegenerative disorder. Neuropediatrics. (2021) 52(2):126–32. doi: 10.1055/s-0040-1715628
Keywords: TPK1, TPK deficiency, thiamine pyrophosphokinase deficiency, outcome predictors, literature review
Citation: Zhao D, Liu M, Jiang H, Song T, Xu C, Duan X, Duan R, Xu H, Liu Z and Fang F (2023) Thiamine pyrophosphokinase deficiency: report of two Chinese cases and a literature review. Front. Pediatr. 11:1173787. doi: 10.3389/fped.2023.1173787
Received: 25 February 2023; Accepted: 24 July 2023;
Published: 9 August 2023.
Edited by:
Sunita Bijarnia-Mahay, Sir Ganga Ram Hospital, IndiaReviewed by:
Juan Dario Ortigoza-Escobar, Sant Joan de Déu Hospital, Spain© 2023 Zhao, Liu, Jiang, Song, Xu, Duan, Duan, Xu, Liu and Fang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Fang ZmFuZ2ZhbmdAYmNoLmNvbS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.