 Rebecca Hetrick
Rebecca Hetrick Melissa Oliver*
Melissa Oliver*
- Division of Pediatric Rheumatology, Indiana University School of Medicine, Indianapolis, IN, United States
Autoinflammatory bone disorders are a group of diseases characterized by sterile osteomyelitis. This includes chronic nonbacterial osteomyelitis and the monogenic forms, Majeed syndrome and deficiency of the interleukin-1 receptor antagonist. These disorders result from innate immune system dysregulation and cytokine imbalance that triggers inflammasome activation causing downstream osteoclastogenesis and excessive bone remodeling. In this review, we will summarize the immunopathogenesis of pediatric autoinflammatory bone diseases with a special focus on the genetics and inborn errors of immunity, while briefly touching on the clinical manifestations and management of each disease as well as areas for future research.
Introduction
Autoinflammatory bone disorders are characterized by sterile bone inflammation that results from innate immune system dysregulation and cytokine imbalance, leading to NLRP3 inflammasome overactivation. This results in pathogenic osteoclastogenesis with excessive bone turnover and bone formation. This review will focus on the genetic mechanisms and inborn errors of immunity that drive sterile bone inflammation in these disorders. These mechanisms provide a framework for understanding the clinical manifestations and management of each disease, as well as potential areas for future research.
Nomenclature
Autoinflammatory bone disorders (ABDs) can be classified as sporadic or monogenic. Sporadic forms of the disease were previously known as chronic recurrent multifocal osteomyelitis (CRMO). However, with increased recognition of unifocal and non-recurrent presentations, the broader term chronic nonbacterial osteomyelitis (CNO) is preferred. Synovitis, Acne, Pustulosis, Hyperostosis, and Osteitis (SAPHO) syndrome also falls within the category of sporadic ABDs. SAPHO tends to present in adults and may represent a part of the CNO spectrum or represent a unique entity within the adult population (1, 2). Majeed syndrome and deficiency of the interleukin-1 receptor antagonist (DIRA) are monogenic ABDs that result from homozygous mutations in the disease-associated gene. Pyogenic arthritis, pyoderma gangrenosum and acne (PAPA), a monogenic autoinflammatory syndrome that can present with sterile bone inflammation, will be briefly discussed (3).
Epidemiology
Despite increased awareness of CNO over the last decade, the disease remains relatively rare. Surveillance data from 2006 to 2008 in Germany estimated an annual incidence of 0.45 per 100,000 children but more recent data suggest a higher incidence (4, 5). Aden et al. demonstrated an increase in the incidence of CNO cases from 8 per million children from 2005 to 2015 compared to 23 per million children from 2016 to 2019 (6).
CNO is typically diagnosed in children and adolescents but can occur in adulthood. In contrast, SAPHO syndrome presents more in adults but can also manifest in children and adolescents. CNO appears more prevalent in females than males with an average age of disease onset occurring between 9 and 11 years of age (6–14). DIRA, Majeed and PAPA are exceptionally rare with a handful of case reports and case series available. DIRA presents at birth or within the first few weeks of life, though intrauterine onset has been described (15, 16). Patients with Majeed present in early childhood most commonly before the age of 2 years (17). PAPA can present in early to late childhood with adult onset also described (18, 19).
Pathogenesis and genetics
Overview
By definition, autoinflammatory diseases are characterized by increased systemic inflammation and an absence of autoantibodies or antigen-specific T cells. In ABDs, research shows that cytokine imbalance and innate immune system dysregulation lead to impaired osteoclastogenesis with increased osteoclastic activation and bone remodeling. This causes the development of sterile bone inflammation. through aberrant activation of the NLRP3 inflammasome, which is an intracellular multi-protein complex involved in host innate immune defenses. In response to microbial infection and/or cellular damage, the NLRP3 inflammasome becomes activated, ultimately leading to secretion of pro-inflammatory cytokines, primarily IL-1 β, which stimulates osteoclast formation (20, 21).
The pathogenesis of CNO is likely multifactorial. An infectious trigger has been investigated but a clear link has yet to be identified. Bone cultures growing microorganisms Propionibacterium acnes, Mycoplasma and Staphylococcus aureus and a partial disease response to azithromycin has been reported (22–29). However, Girschick et al. evaluated this association and found no evidence of bacterial infection in bone biopsy of pediatric CNO patients, suggesting that positive microbial cultures may represent contaminants (23). Whether or not a preceding infection or microbial exposure is involved in the onset of immune dysregulation requires further exploration. Nevertheless, most experts agree that antibiotics alone are insufficient to manage CNO (7, 13, 23, 30).
Inborn errors of immunity and other genetics likely also contribute to the pathogenesis of CNO. Mutations within the NLRP3 inflammasome are associated with ABDs, with specific mutations involving the interleukin (IL)-1 receptor antagonist gene and lipin-2 gene implicated in the monogenic forms of CNO (21, 31). These mutations lead to increased production of the cytokine IL-1β and other pro-inflammatory mediators, which are involved in the pathogenesis of sterile bone inflammation (15). We will further explore the genetics and immunopathogenesis involved in ABDs (summarized in Table 1).
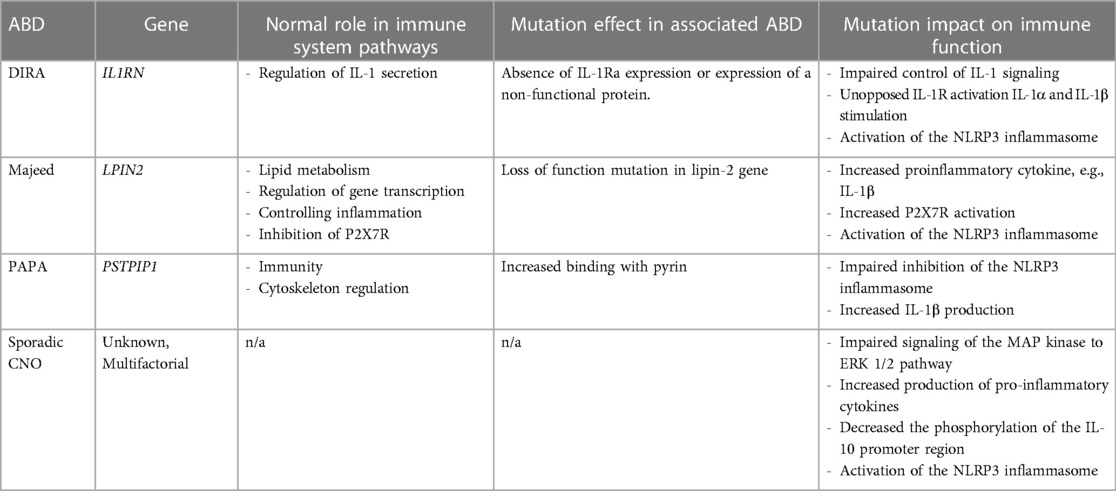
Table 1. Summary of ABDs, associated genetic mutation, and impact on immune function.
Monogenic CNO
IL1RN
Cytokine IL-1 is a key mediator of the innate immune system's inflammatory response (21, 31). IL-1 exists in two forms, IL-1α and IL-1β, both of which are pro-inflammatory cytokines. IL-1 signals through the IL-1 receptor (IL-1R) and IL-1R antagonist (IL-1Ra) regulates IL-1 activity by blocking binding of IL-1 to IL-1R. IL-1β secretion occurs via activated caspase-1, the IL-1 converting enzyme, within the multi-protein complex NLRP3 inflammasome. Within the inflammasome, caspase-1 cleaves pro-interleukin-1β to the biologically active form, IL-1β (32–34). IL-1β induces the expression of genes involved in the acute inflammatory response and fever control. Overproduction of IL-1β has been implicated as the driving force in the pathogenesis of many autoinflammatory disorders (21, 31). Mutations in the IL-1Ra gene (IL1RN) can either cause an absence of IL-1Ra expression or expression of a non-functional protein. This results in impaired control of IL-1 signaling, unopposed IL-1R activation and IL-1α and IL-1β hyperresponsiveness and aberrant inflammatory response (15). Patients with DIRA, one of the monogenic forms of CNO, have mutations in IL1RN (15, 35).
LPIN2
Lipin-2 gene (LPIN2) encodes the protein called lipin-2. Lipin proteins have important roles in lipid metabolism and gene expression as transcriptional co-regulators in adipogenesis. They have also been implicated in controlling inflammation through maintenance of cellular membrane cholesterol levels and adipocyte inflammatory gene expression. LPIN2 is found in the liver, intestines and white blood cells (36). LPIN2 is a phosphatidate phosphatase (PAPs) and loss of PAP activity causes increased inflammation (36, 37). Mutations in LPIN2 lead to cytokine imbalances and deficiency of LPIN2 creates an increase in pro-inflammatory cytokines (IL-1β, IL-6, IFN-γ and TNFα). With reduced or deficient lipin-2 expression in macrophages, increased activation of the NLRP3 inflammasome triggered by the innate immune response results in IL-1β overproduction (36, 38). Lorden et al. has also described lipin-2 as a negative innate immune system regulator through its interaction with the P2X7 receptor (P2X7R) (37). P2X7R is expressed by most immune cells and induces a variety of cellular response, including inflammation, cell proliferation and death, and phagocytosis. Disruptions in the binding of this receptor on macrophages trigger inflammasome activation. LPIN2 may inhibit the activation and sensitization of the P2X7R. A deficiency of lipin-2 protein allows for increased activation of this receptor which downstream activates the NLRP3 inflammasome (37).
A loss of function mutation in LPIN2 has been found in affected individuals with Majeed Syndrome, another rare monogenic form of CNO. Five different mutations in the LPIN2 gene have been reported in consanguineous families. Most patients are homozygous for a nonsense or splice mutation in LPIN2 gene, although one mutation is reported as a missense mutation (Ser734Leu) (17). While the link between lipin-2 and inflammasome regulation has been established, further research is needed to understand the preferential target on bone that produces the clinical phenotype in Majeed Syndrome.
Other genetic associations
Sporadic CNO lacks a direct genetic association. However, a genetic predisposition may in the risk of developing sterile bone inflammation when combined with other factors.
The Pombe Cdc15 homology (PCH) family is a group of proteins involved in immunity and cytoskeleton regulation (39). Proline-serine-threonine phosphatase-interacting protein (PSTPIP)1 and PSTPIP2 proteins are part of the PCH family and are preferentially expressed in the hematopoietic system. Mutations in these proteins have been associated with autoinflammatory disorders (39, 40). Mutations in PSTPIP1 cause pyogenic arthritis, pyoderma gangrenosum, and acne (PAPA) syndrome in humans, while an autosomal recessive mutation in PSTPIP2 has been implicated in the development of multifocal osteomyelitis in mouse models (the cmo mouse) (39–41).
PSTPIP1 mutations can lead to increased binding with pyrin, a protein produced on monocytes involved in the apoptotic and inflammatory signaling pathways. This enhanced interaction of pyrin and PSTPIP1 results in impaired inhibition of the inflammasome and increased IL-1β production in peripheral mononuclear cells (39, 42). PSTPIP1 mutations specifically in T lymphocytes attenuate the phosphorylation of extracellular signal regulated kinase (ERK)1 and ERK2, which are mitogen-activated protein kinases involved in intracellular signal transduction and priming the inflammasome through lipopolysaccharide activation (43, 44). Impairments in the ERK1/2 pathway have also been implicated in the pathogenesis of sporadic CNO, which is discussed later in this review (45, 46).
The PSTPIP2 gene is located on chromosome 18 in both mice and humans and is selectively expressed in macrophages. Disruption of the PSTPIP2 protein creates excessive macrophage accumulation and increased production of pro-inflammatory mediators, such as chemokine macrophage inflammatory protein-1α (MIP-1α) and cytokine IL-6 (48). The loss of regulation of macrophages and inflammatory mediators promotes osteoclast and osteoblast activation (49). A missense mutation, specifically L98P, has been found to alter the structure and/or function of PSTPIP2 causing sterile bone inflammation in mice that mimics CNO seen in humans (41, 47, 48). The paws of the cmo mouse have elevated levels of pro-inflammatory cytokines and chemokines, particularly IL-1β. The cmo mouse will then develop sterile osteomyelitis in their tails and paws beginning at 4 to 6 weeks (41). While this mutation in PSTPIP2 is implicated in the disease pathogenesis of CNO in mice, its role in human CNO is unclear.
FBLIM1 is another gene where mutations have been implicated in the development of sterile bone inflammation. This encodes filamin-binding LIM protein 1 (FBLP-1), which is involved in bone remodeling through the regulation of receptor activator of nuclear factor (NF)-kβ (RANK) ligand (RANKL) activation via the ERK1/2 phosphorylation pathway mentioned earlier with mutations involving PSTPIP1. RANK/RANKL signaling controls normal osteoclast function and is important for bone homeostasis (50, 51). RANKL binds to its receptor RANK on osteoclast precursors resulting in the activation of multiple signaling pathways that lead to osteoclast differentiation and increased bone turnover (50). Using whole exome sequencing, Cox et al, identified mutations in the FBLIM1 gene in two children with CNO and psoriasis. It is postulated that when FBLP1 is absent or dysfunctional, this leads to increased ERK1/2 phosphorylation and increased RANKL production that causes osteoclast activation, bone resorption, and inflammasome activation (52).
Further supporting a possible genetic susceptibility for CNO, Golla et al. genotyped CNO patients and their parents and found a significant association with a rare allele of marker D18S60 on chromosome 18q21.3–22 (53). Another researcher, Charras, et al., performed whole exome sequencing in families with CNO and found rare damaging heterozygous variants within the P2X7R sequence, which may play a role in CNO development and severity of the disease (54). Both need further investigations, but these suggest there may be other genes that could predispose individuals to develop CNO.
Sporadic CNO
Although lacking a single responsible gene, the sterile bone inflammation in sporadic CNO involves many of the same pathways and cellular machinery as the monogenic forms. It develops from a combination of impaired innate immune responses and an imbalance of cytokine expression (21, 55). Monocytes from sporadic CNO patients fail to express the immune regulatory cytokines IL-10 and IL-19 because of impaired signaling of the MAP kinase (MAPK) to theERK 1/2 phosphorylation pathway. Other environmental stress- or mitogen response-signaling pathways, the c-Jun N-terminal kinase (JNK) and p38 MAPK pathways, are unaffected. Thus, this imbalance leads to increased production of pro-inflammatory cytokines (IL-1β, IL-6, TNFα, monocyte chemoattractant protein 1, MIP1-β) (45, 46). Additionally, there is a decreased phosphorylation of the IL-10 promoter region due to genetic polymorphisms resulting in further impaired the regulatory cytokine IL-10 expression. Of note, this reduced IL-10 expression may be related to FBLIM1's involvement in disease pathogenesis mentioned earlier because IL-10 mediates transcription factor Signal Transducer and Activator of Transcription (STAT)3 which increases the gene expression of FBLIM1 (45). Recognition of intracellular danger signals (PRR, TLR, NLR) by CNO monocytes further amplifies the NLRP3 inflammasome activity (33). Ultimately, the increased activation of NLRP3 inflammasome causes increased production of IL-1β which leads to pathogenic osteoclastogenesis via enhanced RANK/RANKL interaction on osteoclast precursor cells, inducing osteoclast differentiation and activation and excessive bone turnover and bone formation (56, 57).
Clinical presentation
DIRA
DIRA syndrome (OMIM #612852) is an autosomal recessive disorder characterized by neonatal onset of systemic inflammation, sterile bone inflammation, and pustular skin lesions. Many neonates present to medical attention with fevers and a septic-like picture that mimics serious bacterial infections (15, 58). However, DIRA represents a clinical spectrum and later presentations have been described (59, 60). Other common findings in DIRA include oral mucosal lesions, nail changes, conjunctival injection, and hepatosplenomegaly (15). If unrecognized and untreated, DIRA carries a significant risk of mortality (58). Delays in treatment can also lead to severe sequelae such as failure to thrive, skeletal deformities, and interstitial lung disease (15, 58).
Majeed
Majeed syndrome (OMIM #609628) is an autosomal recessive disorder first described in 3 related children presenting with sterile bone inflammation and congenital dyserythropoietic anemia (CDA) (61). Since these first cases, 21 additional affected individuals have been described. Sterile bone inflammation and microcytic dyserythropoietic anemia are the most prominent clinical features, occurring in 91% of affected individuals. Skin manifestations, primarily neutrophilic dermatoses, are considered characteristic of the disease but only appear in 14% (62). Other common findings include elevated inflammatory markers (88%), recurrent fevers (46%), failure to thrive (38%), hepatosplenomegaly (30%), and neutropenia (13%) (17).
PAPA
Pyogenic Arthritis, Pyoderma Gangrenosum, and Acne syndrome (OMIM #604416) is an autosomal dominant disorder characterized by aseptic arthritis, pyoderma gangrenosum, and cystic acne. However, only 25% of patients experience the triad of manifestations. Arthritis occurs in most patients and can be erosive, polyarticular or oligoarticular, and typically affects peripheral joints (63). Cystic acne affects an estimated 10%–20%, and fevers accompany disease flares in 27% (18, 19, 63). Sterile osteomyelitis is reported in some cases of PAPA with one series finding sterile bone inflammation in 8% (3, 63, 64).
Sporadic CNO
CNO typically presents with bone pain with or without associated swelling or warmth. CNO is a diagnosis of exclusion without validated diagnostic criteria, and ruling out mimicker diseases, such as infection and malignancy, plays a role in the evaluation. CNO lesions can affect any bone but most commonly occur in the metaphyseal regions of long bones. However, epiphyseal and diaphyseal lesions can be seen, occurring in 37% of patients in one case series (9). Other commonly affected bones include the mandible, clavicles, vertebrae, and bones of the pelvis. Bone involvement can be unifocal or multifocal and symmetric or asymmetric (6–14, 64).
Many CNO patients experience inflammation elsewhere in the body, most notably in the joints, skin, and gut (6–14). Joint involvement may be seen in up to 40% of CNO patients and is typically monoarticular or oligoarticular, but polyarticular disease can occur (9, 10). Likewise, arthritis in CNO prefers joints adjacent to boney lesions but can also occur at distant sites (10, 13). Common associated skin conditions include psoriasis, palmoplantar pustulosis, and severe acne, occurring in anywhere from 10%–24% of CNO patients. Comorbid inflammatory bowel disease is estimated to occur in 3%–8% of CNO patients (6–14).
SAPHO presents with similar bone pain and skeletal findings as CNO except there is a predilection for the sternoclavicular area. Patients with SAPHO have more cutaneous involvement, particularly palmoplantar pustulosis(66).
Diagnosis
Laboratory evaluation
There are no specific lab findings or pathognomonic autoantibodies for ABDs (21, 67). Monogenic ABDs typically have evidence of systemic inflammation with elevated c-reactive protein (CRP) and erythrocyte sedimentary rate (ESR) and/or leukocytosis (15, 17–19). In sporadic CNO, lab evidence of systemic inflammation may or may not be present (6–14).
Imaging evaluation
Radiologic evaluation plays a critical role in detecting osteitis and evaluating disease extent. Plain radiographs are generally first-line for the evaluation of bone pain in children. However, they have low sensitivity for early disease findings. MRI has emerged as the gold standard for the evaluation of osteitis (68, 69). MRI can demonstrate early disease findings such as bony edema or altered diffusion capacity on diffusion-weighted imaging. Other findings of active lesions include periosteal reaction, hyperostosis, and surrounding soft tissue inflammation (65). MRI can also reveal disease morbidities such as pathologic fractures, vertebral compression fractures, or growth plate disruption (70). Whole-body MRI is preferred given the presence of clinically silent lesions in up to 29% of patients (12, 69). While bone scintigraphy may be used when whole-body MRI is unavailable, this modality is less sensitive and involves radiation exposure (9, 68).
Bone biopsy
Bone biopsy remains an important diagnostic tool in certain cases. Clinicians may opt to forgo bone biopsy in patients with a typical clinical presentation and compatible imaging findings. However, certain clinical scenarios, such as the presence of solitary lesions, cytopenia, or excessively elevated inflammatory markers, may warrant biopsy if infection or malignancy remains on the differential (71). Histopathology of bone samples can vary depending on the stage of the disease. Early lesions are characterized by the presence of neutrophils and monocytes (72, 73). Older lesions are characterized by the presence of lymphocytes and plasma cells and often demonstrate chronic inflammatory change with some degree of fibrosis or sclerosis (7, 72, 73).
Management of autoinflammatory bone diseases
Treatment of monogenic ABDs appears to correlate with the understood pathophysiology of these syndromes. Both anakinra, a recombinant human IL-1Ra, and rilonacept, a recombinant fusion protein that binds to both IL-1β and IL-1apha, are recommended first-line agents for DIRA (74, 75). IL-1β antagonist canakinumab does not appear as consistently efficacious as these agents, possibly due to ongoing IL-1α activity in these patients (60, 75, 76). Similarly, Majeed Syndrome appears to response well to anti-IL-1 activity, with reports of efficacy with anakinra and canakinumab (77, 78). However, the management of PAPA is much more variable. Good response has been reported to corticosteroids, anakinra, canakinumab, TNF inhibitors, and tocilizumab (63).
Therapies for sporadic CNO either mediate pro-inflammatory cytokine activity or target osteoclast activity. Treatment of sporadic CNO has advanced in recent years with the development of the Childhood Arthritis and Rheumatology Research Alliance (CARRA) consensus treatment plan (68). First line therapy for CNO patients is typically non-steroidal anti-inflammatory drug (NSAID) therapy unless spine involvement is present. NSAIDs inhibit the activity of cyclooxygenases (COX). These enzymes convert arachidonic acid into prostaglandins, which are required for osteoclast activation (56, 79).
Unfortunately, only about 40% of sporadic CNO patients achieve disease control with NSAIDs monotherapy. Many require escalation to biologic or non-biologic disease-modifying antirheumatic drug (DMARD) therapy and/or bisphosphonates (9, 14). Non-biologic DMARDs, such as methotrexate and sulfasalazine, provide immunomodulatory effects through multiple mechanisms, but the direct therapeutic mechanism of these agents in sporadic CNO remains unclear (79). Biologic DMARDs function by disabling or blocking the activity of specific pro-inflammatory cytokines. TNF-α inhibitors are the most common biologic DMARDs used in sporadic CNO. These medications inactivate TNF-α, a very potent pro-inflammatory cytokine found to be elevated in the monocytes and serum of patients with sporadic CNO (45, 56). Bisphosphonates are preferred in patients with spinal lesions and often used in patients with NSAID-refractory disease (68). Bisphosphonates work through a variety of mechanisms to inhibit osteoclast activity and impair osteoclast function, but the exact therapeutic mechanism in sporadic CNO remains unclear (79).
Lastly, given the implication of the IL-1 pathway in the pathogenesis of CNO, modulation of this pathway has been proposed as a promising therapy. However, treatment of sporadic CNO with anti-IL-1 therapy has shown mixed results (80–82).
Future directions
Despite the major advances made over the last twenty years, there are still many unanswered questions in ABDs research. CNO remains a diagnosis of exclusion. With no specific biomarkers, patients often experience diagnostic and treatment delays. A group of international experts and CNO patients and families are working to develop the first classification criteria for pediatric CNO to aid in the earlier identification of these patients (83, 84).
Unfortunately, most data on ABDs comes from case reports and series or retrospective cohort studies. The CRMO/CNO workgroup within CARRA has established an international prospective disease registry (CHronic nonbacterial Osteomyelitis International Registry (CHOIR)) for patients with chronic nonbacterial osteomyelitis (CNO). From the CHOIR registry, a prospective observation study is currently underway to determine the effectiveness of the consensus treatment plans previously developed by the CARRA workgroup. The registry will aid researchers in conducting comparative effectiveness trials (85). At this time, the field of ABDs lacks validated outcomes measures that would facilitate comparative effectiveness of different therapies. The OMERACT (Outcome Measures in Rheumatoid Arthritis Clinical Trials) CNO/SAPHO working group was established in 2019 to develop a core outcome measurement set through a data driven and consensus process for CNO and SAPHO for all clinical trials (86).
Conclusion
Autoinflammatory bone disorders constitute a heterogeneous group of rare diseases with both sporadic and monogenic forms. Extraosseous manifestations, most commonly involving the skin, often occur. Dysregulation of the innate immune system and cytokine imbalance contribute to disease pathogenesis. Critical areas of future research include the development of diagnostic and classification criteria, enrollment in large prospective cohorts, and creation of standardized outcomes measures.
Author contributions
RH: review concept and design, literature review, drafting and revision of the manuscript. MO: review concept and design, literature review, drafting and revision of the manuscript and supervision. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Letts M, Davidson D, Birdi N, Joseph M. The SAPHO syndrome in children: a rare cause of hyperostosis and osteitis. J Pediatr Orthop. (1999) 19(3):297–300. PMID: 10344310.10344310
2. Lenert A, Ferguson PJ. Comparing children and adults with chronic nonbacterial osteomyelitis. Curr Opin Rheumatol. (2020) 32(5):421–6. doi: 10.1097/BOR.0000000000000734
3. Caorsi R, Picco P, Buoncompagni A, Martini A, Gattorno M. Osteolytic lesion in PAPA syndrome responding to anti-interleukin 1 treatment. J Rheumatol. (2014) 41(11):2333–4. doi: 10.3899/jrheum.140060
4. Jansson AF, Grote V, ESPED Study Group. Nonbacterial osteitis in children: data of a German incidence surveillance study. Acta Paediatr. (2011) 100(8):1150–7. doi: 10.1111/j.1651-2227.2011.02205.x
5. Schnabel A, Range U, Hahn G, Siepmann T, Berner R, Hedrich CM. Unexpectedly high incidences of chronic non-bacterial as compared to bacterial osteomyelitis in children. Rheumatol Int. (2016) 36(12):1737–45. doi: 10.1007/s00296-016-3572-6
6. Aden S, Wong S, Yang C, Bui T, Higa T, Scheck J, et al. Increasing cases of chronic nonbacterial osteomyelitis in children: a series of 215 cases from a single tertiary referral center. J Rheumatol. (2022) 49(8):929–34. doi: 10.3899/jrheum.210991
7. Jansson A, Renner ED, Ramser J, Mayer A, Haban M, Meindl A, et al. Classification of non-bacterial osteitis: retrospective study of clinical, immunological and genetic aspects in 89 patients. Rheumatology. (2007) 46(1):154–60. doi: 10.1093/rheumatology/kel190
8. Beck C, Morbach H, Beer M, Stenzel M, Tappe D, Gattenlöhner S, et al. Chronic nonbacterial osteomyelitis in childhood: prospective follow-up during the first year of anti-inflammatory treatment. Arthritis Res Ther. (2010) 12(2):1. doi: 10.1186/ar2992
9. Girschick H, Finetti M, Orlando F, Schalm S, Insalaco A, Ganser G, et al. The multifaceted presentation of chronic recurrent multifocal osteomyelitis: a series of 486 cases from the Eurofever international registry. Rheumatology. (2018) 57(7):1203–11. doi: 10.1093/rheumatology/key058
10. Borzutzky A, Stern S, Reiff A, Zurakowski D, Steinberg EA, Dedeoglu F, et al. Pediatric chronic nonbacterial osteomyelitis. Pediatrics. (2012) 130(5):e1190–7. doi: 10.1542/peds.2011-3788
11. Wipff J, Costantino F, Lemelle I, Pajot C, Duquesne A, Lorrot M, et al. A large national cohort of French patients with chronic recurrent multifocal osteitis. Arthritis Rheumatol. (2015) 67(4):1128–37. doi: 10.1002/art.39013
12. Roderick MR, Shah R, Rogers V, Finn A, Ramanan AV. Chronic recurrent multifocal osteomyelitis (CRMO)–advancing the diagnosis. Pediatr Rheumatol. (2016) 14(1):1–5. doi: 10.1186/s12969-016-0109-1
13. Huber AM, Lam PY, Duffy CM, Yeung RS, Ditchfield M, Laxer D, et al. Chronic recurrent multifocal osteomyelitis: clinical outcomes after more than five years of follow-up. J Pediatr. (2002) 141(2):198–203. doi: 10.1067/mpd.2002.126457
14. Schnabel A, Range U, Hahn G, Berner R, Hedrich CM. Treatment response and longterm outcomes in children with chronic nonbacterial osteomyelitis. J Rheumatol. (2017) 44(7):1058–65. doi: 10.3899/jrheum.161255
15. Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, et al. An autoinflammatory disease with deficiency of the interleukin-1–receptor antagonist. N Engl J Med. (2009) 360(23):2426–37. doi: 10.1056/NEJMoa0807865
16. Altiok E, Aksoy F, Perk Y, Taylan F, Kim PW, Ilıkkan B, et al. A novel mutation in the interleukin-1 receptor antagonist associated with intrauterine disease onset. Clin Immunol. (2012) 145(1):77–81. doi: 10.1016/j.clim.2012.08.003
17. Ferguson PJ, El-Shanti H. Majeed syndrome: a review of the clinical, genetic and immunologic features. Biomolecules. (2021) 11(3):367. doi: 10.3390/biom11030367
18. Lindor NM, Arsenault TM, Solomon H, Seidman CE, McEvoy MT. A new autosomal dominant disorder of pyogenic sterile arthritis, pyoderma gangrenosum, and acne: PAPA syndrome. Mayo Clin Proc. (1997) 72(7):611–5. doi: 10.1016/S0025-6196(11)63565-9
19. Yeon HB, Lindor NM, Seidman JG, Seidman CE. Pyogenic arthritis, pyoderma gangrenosum, and acne syndrome maps to chromosome 15q. Am J Hum Genet. (2000) 66(4):1443–8. doi: 10.1086/302866
20. Hofmann SR, Kubasch AS, Range U, Laass MW, Morbach H, Girschick HJ, et al. Serum biomarkers for the diagnosis and monitoring of chronic recurrent multifocal osteomyelitis (CRMO). Rheumatol Int. (2016) 36:769–79. doi: 10.1007/s00296-016-3466-7
21. Morbach H, Hedrich CM, Beer M, Girschick HJ. Autoinflammatory bone disorders. Clin Immunol. (2013) 147(3):185–96. doi: 10.1016/j.clim.2012.12.012
22. Bj0rksten B, Boquist LE. Histopathological aspects of chronic recurrent multifocal osteomyelitis. J Bone Joint Surg Br. (1980) 62(3):376–80. doi: 10.1302/0301-620X.62B3.7410472
23. Girschick HJ, Huppertz HI, Harmsen D, Krauspe R, Müller-Hermelink HK, Papadopoulos T. Chronic recurrent multifocal osteomyelitis in children: diagnostic value of histopathology and microbial testing. Hum Pathol. (1999) 30(1):59–65. doi: 10.1016/S0046-8177(99)90301-5
24. Assmann G, Kueck O, Kirchhoff T, Rosenthal H, Voswinkel J, Pfreundschuh M, et al. Efficacy of antibiotic therapy for SAPHO syndrome is lost after its discontinuation: an interventional study. Arthritis Res Ther. (2009) 11(5):1–8. doi: 10.1186/ar2812
25. Schilling F, Wagner AD. Azithromycin: an anti-inflammatory effect in chronic recurrent multifocal osteomyelitis? A preliminary report. Z Rheumatol. (2000) 59(5):352–3. doi: 10.1007/s003930070059
26. Edlund E, Johnsson U, Lidgren LA, Pettersson H, Sturfelt G, Svensson B, et al. Palmoplantar pustulosis and sternocostoclavicular arthro-osteitis. Ann Rheum Dis. (1988) 47(10):809–15. doi: 10.1136/ard.47.10.809
27. Colina M, Monaco AL, Khodeir M, Trotta F. Propionibacterium acnes and SAPHO syndrome: a case report and literature. Clin Exp Rheumatol. (2007) 25:457–60. PMID: 17631745.17631745
28. Hummell DS, Anderson SJ, Wright PF, Cassell GH, Waites KB. Chronic recurrent multifocal osteomyelitis: are mycoplasmas involved? N Engl J Med. (1987) 317(8):510–1. PMID: 3614302.3614302
29. King SM, Laxer RM, Manson D, Gold RO. Chronic recurrent multifocal osteomyelitis: a noninfectious inflammatory process. Pediatr Infect Dis J. (1987) 6(10):907–10. doi: 10.1097/00006454-198710000-00009
30. Schultz C, Holterhus PM, Seidel A, Jonas S, Barthel M, Kruse K, et al. Chronic recurrent multifocal osteomyelitis in children. Pediatr Infect Dis J. (1999) 18(11):1008–13. doi: 10.1097/00006454-199911000-00015
31. Goldbach-Mansky R. Immunology in clinic review series; focus on autoinflammatory diseases: update on monogenic autoinflammatory diseases: the role of interleukin (IL)-1 and an emerging role for cytokines beyond IL-1. Clin Exp Immunol. (2012) 167(3):391–404. doi: 10.1111/j.1365-2249.2011.04533.x
32. Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunol Rev. (2015) 265(1):6–21. doi: 10.1111/imr.12296
33. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. (2019) 20(13):3328. doi: 10.3390/ijms20133328
34. Kaneko N, Kurata M, Yamamoto T, Morikawa S, Masumoto J. The role of interleukin-1 in general pathology. Inflamm Regen. (2019) 39:1–6. doi: 10.1186/s41232-019-0101-5
35. Reddy S, Jia S, Geoffrey R, Lorier R, Suchi M, Broeckel U, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med. (2009) 360(23):2438–44. doi: 10.1056/NEJMoa0809568
36. Zhang P, Reue K. Lipin proteins and glycerolipid metabolism: roles at the ER membrane and beyond. Biochim Biophys Acta Biomembr. (2017) 1859(9):1583–95. doi: 10.1016/j.bbamem.2017.04.007
37. Lordén G, Sanjuán-García I, de Pablo N, Meana C, Alvarez-Miguel I, Pérez-García MT, et al. Lipin-2 regulates NLRP3 inflammasome by affecting P2X7 receptor activation. J Exp Med. (2017) 214(2):511–28. doi: 10.1084/jem.20161452
38. Valdearcos M, Esquinas E, Meana C, Peña L, Gil-de-Gómez L, Balsinde J, et al. Lipin-2 reduces proinflammatory signaling induced by saturated fatty acids in macrophages. J Biol Chem. (2012) 287(14):10894–904. doi: 10.1074/jbc.M112.342915
39. Chitu V, Stanley ER. Pombe Cdc15 homology (PCH) proteins: coordinators of membrane–cytoskeletal interactions. Trends Cell Biol. (2007) 17(3):145–56. doi: 10.1016/j.tcb.2007.01.003
40. Cassel SL, Janczy JR, Bing X, Wilson SP, Olivier AK, Otero JE, et al. Inflammasome-independent IL-1β mediates autoinflammatory disease in Pstpip2-deficient mice. Proc Natl Acad Sci USA. (2014) 111(3):1072–7. doi: 10.1073/pnas.1318685111
41. Ferguson PJ, Bing X, Vasef MA, Ochoa LA, Mahgoub A, Waldschmidt TJ, et al. A missense mutation in pstpip2 is associated with the murine autoinflammatory disorder chronic multifocal osteomyelitis. Bone. (2006) 38(1):41–7. doi: 10.1016/j.bone.2005.07.009
42. Stutz A, Kolbe CC, Stahl R, Horvath GL, Franklin BS, van Ray O, et al. NLRP3 Inflammasome assembly is regulated by phosphorylation of the pyrin domain. J Exp Med. (2017) 214(6):1725–36. doi: 10.1084/jem.20160933
43. Yang H, Reinherz EL. CD2BP1 Modulates CD2-dependent T cell activation via linkage to protein tyrosine phosphatase (PTP)-PEST. J Immunol. (2006) 176(10):5898–907. doi: 10.4049/jimmunol.176.10.5898
44. Ghonime MG, Shamaa OR, Das S, Eldomany RA, Fernandes-Alnemri T, Alnemri ES, et al. Inflammasome priming by lipopolysaccharide is dependent upon ERK signaling and proteasome function. J Immunol. (2014) 192(8):3881–8. doi: 10.4049/jimmunol.1301974
45. Hofmann SR, Schwarz T, Möller JC, Morbach H, Schnabel A, Rösen-Wolff A, et al. Chronic non-bacterial osteomyelitis is associated with impaired Sp1 signaling, reduced IL10 promoter phosphorylation, and reduced myeloid IL-10 expression. Clin Immunol. (2011) 141(3):317–27. doi: 10.1016/j.clim.2011.08.012
46. Hofmann SR, Morbach H, Schwarz T, Rösen-Wolff A, Girschick HJ, Hedrich CM. Attenuated TLR4/MAPK signaling in monocytes from patients with CRMO results in impaired IL-10 expression. Clin Immunol. (2012) 145(1):69–76. doi: 10.1016/j.clim.2012.07.012
47. Grosse J, Chitu V, Marquardt A, Hanke P, Schmittwolf C, Zeitlmann L, et al. Mutation of mouse Mayp/Pstpip2 causes a macrophage autoinflammatory disease. Blood. (2006) 107(8):3350–8. doi: 10.1182/blood-2005-09-3556
48. Chitu V, Ferguson PJ, De Bruijn R, Schlueter AJ, Ochoa LA, Waldschmidt TJ, et al. Primed innate immunity leads to autoinflammatory disease in PSTPIP2-deficient cmo mice. Blood. J Am Soc Hematol. (2009) 114(12):2497–505. doi: 10.1182/blood-2009-02-204925
49. Lukens JR, Gross JM, Calabrese C, Iwakura Y, Lamkanfi M, Vogel P, et al. Critical role for inflammasome-independent IL-1β production in osteomyelitis. Proc Natl Acad Sci USA. (2014) 111(3):1066–71. doi: 10.1073/pnas.1318688111
50. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. (2003) 423(6937):337–42. doi: 10.1038/nature01658
51. Xiao G, Cheng H, Cao H, Chen K, Tu Y, Yu S, et al. Critical role of filamin-binding LIM protein 1 (FBLP-1)/migfilin in regulation of bone remodeling. J Biol Chem. (2012) 287(25):21450–60. doi: 10.1074/jbc.M111.331249
52. Cox AJ, Darbro BW, Laxer RM, Velez G, Bing X, Finer AL, et al. Recessive coding and regulatory mutations in FBLIM1 underlie the pathogenesis of chronic recurrent multifocal osteomyelitis (CRMO). PLoS One. (2017) 12(3):e0169687. doi: 10.1371/journal.pone.0169687
53. Golla A, Jansson A, Ramser J, Hellebrand H, Zahn R, Meitinger T, et al. Chronic recurrent multifocal osteomyelitis (CRMO): evidence for a susceptibility gene located on chromosome 18q21. 3-18q22. Eur J Hum Genet. (2002) 10(3):217–21. doi: 10.1038/sj.ejhg.5200789
54. Charras A, Hofmann SR, Cox A, Schulze F, Russ S, Hartmann H, et al. 678 Damaging variants in P2X7R associate with Chronic Nonbacterial Osteomyelitis (CNO).
55. Scianaro R, Insalaco A, Bracci Laudiero L, De Vito R, Pezzullo M, Teti A, et al. Deregulation of the IL-1β axis in chronic recurrent multifocal osteomyelitis. Pediatr Rheumatol. (2014) 12(1):1–6. doi: 10.1186/1546-0096-12-30
56. Nakashima T, Takayanagi H. Osteoclasts and the immune system. J Bone Miner Metab. (2009) 27:519–29. doi: 10.1007/s00774-009-0089-z
57. Nakashima T, Takayanagi H. Osteoimmunology: crosstalk between the immune and bone systems. J Clin Immunol. (2009) 29:555–67. doi: 10.1007/s10875-009-9316-6
58. Mendonca LO, Malle L, Donovan FX, Chandrasekharappa SC, Montealegre Sanchez GA, Garg M, et al. Deficiency of interleukin-1 receptor antagonist (DIRA): report of the first Indian patient and a novel deletion affecting IL1RN. J Clin Immunol. (2017) 37:445–51. doi: 10.1007/s10875-017-0399-1
59. Sözeri B, Gerçeker-Türk B, Yıldız-Atıkan B, Mir S, Berdeli A. A novel mutation of interleukin-1 receptor antagonist (IL1RN) in a DIRA patient from Turkey: diagnosis and treatment. Turk J Pediatr. (2018) 60(5):588–92. doi: 10.24953/turkjped.2018.05.020
60. Kuemmerle-Deschner JB, Welzel T, Hoertnagel K, Tsiflikas I, Hospach A, Liu X, et al. New variant in the IL1RN-gene (DIRA) associated with late-onset, CRMO-like presentation. Rheumatology. (2020) 59(11):3259–63. doi: 10.1093/rheumatology/keaa119
61. Majeed HA, Al-Tarawna M, El-Shanti H, Kamel B, Al-Khalaileh F. The syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia. Report of a new family and a review. Eur J Pediatr. (2001) 160:705–10. doi: 10.1007/s004310100799
62. Majeed HA, Kalaawi M, Mohanty D, Teebi AS, Tunjekar MF, Al-Gharbawy F, et al. Congenital dyserythropoietic anemia and chronic recurrent multifocal osteomyelitis in three related children and the association with sweet syndrome in two siblings. J Pediatr. (1989) 115(5):730–4. doi: 10.1016/S0022-3476(89)80650-X
63. Wang Y, Wu N, Yu K, Shen M. Case report: pyogenic arthritis, pyoderma gangrenosum, and acne: a single-center experience and literature review. Front Immunol. (2021) 12:735851. doi: 10.3389/fimmu.2021.735851
64. Demidowich AP, Freeman AF, Kuhns DB, Aksentijevich I, Gallin JI, Turner ML, et al. Brief report: genotype, phenotype, and clinical course in five patients with PAPA syndrome (pyogenic sterile arthritis, pyoderma gangrenosum, and acne). Arthritis Rheum. (2012) 64(6):2022–7. doi: 10.1002/art.34332
65. d’Angelo P, de Horatio LT, Toma P, Ording Müller LS, Avenarius D, von Brandis E, et al. Chronic nonbacterial osteomyelitis—clinical and magnetic resonance imaging features. Pediatr Radiol. (2021) 51:282–8. doi: 10.1007/s00247-020-04827-6
66. Rukavina I. SAPHO Syndrome: a review. J Child Orthop. (2015) 9(1):19–27. doi: 10.1007/s11832-014-0627-7
67. Park H, Bourla AB, Kastner DL, Colbert RA, Siegel RM. Lighting the fires within: the cell biology of autoinflammatory diseases. Nat Rev Immunol. (2012) 12(8):570–80. doi: 10.1038/nri3261
68. Zhao Y, Wu EY, Oliver MS, Cooper AM, Basiaga ML, Vora SS, et al. Consensus treatment plans for chronic nonbacterial osteomyelitis refractory to nonsteroidal antiinflammatory drugs and/or with active spinal lesions. Arthritis Care Res (Hoboken). (2018) 70(8):1228–37. doi: 10.1002/acr.23462
69. Hedrich CM, Morbach H, Reiser C, Girschick HJ. New insights into adult and paediatric chronic non-bacterial osteomyelitis CNO. Curr Rheumatol Rep. (2020) 22:1. doi: 10.1007/s11926-020-00928-1
70. Zhao Y, Chauvin NA, Jaramillo D, Burnham JM. Aggressive therapy reduces disease activity without skeletal damage progression in chronic nonbacterial osteomyelitis. J Rheumatol. (2015) 42(7):1245–51. doi: 10.3899/jrheum.141138
71. Zhao Y, Ferguson PJ. Chronic nonbacterial osteomyelitis and chronic recurrent multifocal osteomyelitis in children. Pediatr Clin. (2018) 65(4):783–800. doi: 10.1016/j.pcl.2018.04.003
72. Brandt D, Sohr E, Pablik J, Schnabel A, Kapplusch F, Mäbert K, et al. CD14+ Monocytes contribute to inflammation in chronic nonbacterial osteomyelitis (CNO) through increased NLRP3 inflammasome expression. Clin Immunol. (2018) 196:77–84. doi: 10.1016/j.clim.2018.04.011
73. Hedrich CM, Hofmann SR, Pablik J, Morbach H, Girschick HJ. Autoinflammatory bone disorders with special focus on chronic recurrent multifocal osteomyelitis (CRMO). Pediatr Rheumatol. (2013) 11(1):1. doi: 10.1186/1546-0096-11-47
74. Hallegua DS, Weisman MH. Potential therapeutic uses of interleukin 1 receptor antagonists in human diseases. Ann Rheum Dis. (2002) 61(11):960–7. doi: 10.1136/ard.61.11.960
75. Romano M, Arici ZS, Piskin D, Alehashemi S, Aletaha D, Barron KS, et al. The 2021 EULAR/American college of rheumatology points to consider for diagnosis, management and monitoring of the interleukin-1 mediated autoinflammatory diseases: cryopyrin-associated periodic syndromes, tumour necrosis factor receptor-associated periodic syndrome, mevalonate kinase deficiency, and deficiency of the interleukin-1 receptor antagonist. Ann Rheum Dis. (2022) 81(7):907–21. doi: 10.1136/annrheumdis-2021-221801
76. Del Giudice E, Sota J, Orlando F, Picciano L, Cimaz R, Cantarini L, et al. Off-label use of canakinumab in pediatric rheumatology and rare diseases. Front Med (Lausanne). (2022) 9:998281. doi: 10.3389/fmed.2022.998281
77. Herlin T, Fiirgaard B, Bjerre M, Kerndrup G, Hasle H, Bing X, et al. Efficacy of anti-IL-1 treatment in Majeed syndrome. Ann Rheum Dis. (2013) 72(3):410–3. doi: 10.1136/annrheumdis-2012-201818
78. Al Mosawi Z, Madan W, Al Moosawi B, Sayed AW, Naser H, Fuad AL. Dramatic response of familial majeed syndrome to interleukin-1 antagonist therapy: case report. Arch Rheumatol. (2019) 34(3):352. doi: 10.5606/ArchRheumatol.2019.7267
79. Hofmann SR, Schnabel A, Rösen-Wolff A, Morbach H, Girschick HJ, Hedrich CM. Chronic nonbacterial osteomyelitis: pathophysiological concepts and current treatment strategies. J Rheumatol. (2016) 43(11):1956–64. doi: 10.3899/jrheum.160256
80. Pardeo M, Marafon DP, Messia V, Garganese MC, De Benedetti F, Insalaco A. Anakinra in a cohort of children with chronic nonbacterial osteomyelitis. J Rheumatol. (2017) 44(8):1231–8. doi: 10.3899/jrheum.160690
81. Acierno S, Angrisani F, Marino A, Caporali RF, Cimaz R, Giani T. Canakinumab treatment in a young girl with refractory chronic recurrent multifocal osteomyelitis associated with pyoderma gangrenosum. Int J Rheum Dis. (2022) 25(11):1333–8. doi: 10.1111/1756-185X.14425
82. Eleftheriou D, Gerschman T, Sebire N, Woo P, Pilkington CA, Brogan PA. Biologic therapy in refractory chronic non-bacterial osteomyelitis of childhood. Rheumatology. (2010) 49(8):1505–12. doi: 10.1093/rheumatology/keq122
83. Oliver M, Wu E, Naden R, Hollander M, Ferguson P, Dedeoglu F, et al. OP0342 Identifying candidate items towards the development of classification criteria for chronic nonbacterial osteomyelitis (cno) and chronic recurrent multifocal osteomyelitis (CRMO). Ann Rheum Dis. (2019) 78:254–5.
84. Zhao Y, Naden R, Oliver M, Wang Z, Wu E, Aguiar C, et al. Comparison of clinicopathologic and imaging features between chronic nonbacterial osteomyelitis and its mimickers: a multi-national 450 case-control study. Arth Rheumatol. (2020) 72:2334–5.
85. US National Institutes of Health. ClinicalTrials.gov (2021). Available at: https://clinicaltrials.gov/ct2/show/NCT04725422
86. Oliver M, Jayatilleke A, Wu E, Nuruzzaman F, Aguiar C, Lenert A, et al. Establishing core domain sets for chronic nonbacterial osteomyelitis (CNO) and synovitis, acne, pustulosis, hyperostosis, osteitis (SAPHO): a report from the OMERACT 2020 special interest group. Semin Arthritis Rheum. (2021,) 51(4):957–61. doi: 10.1016/j.semarthrit.2021.05.015
Keywords: autoinflammatory bone disorders, chronic nonbacterial osteomyelitis, chronic recurrent multifocal osteomyelitis, synovitis acne pustulosis hyperostosis osteitis (SAPHO) syndrome, Majeed syndrome, deficiency of the interleukin-1 receptor antagonist
Citation: Hetrick R and Oliver M (2023) Pediatric autoinflammatory bone disorders—a mini review with special focus on pathogenesis and inborn errors of immunity. Front. Pediatr. 11:1169659. doi: 10.3389/fped.2023.1169659
Received: 19 February 2023; Accepted: 17 May 2023;
Published: 5 June 2023.
Edited by:
Mildred Kwan, University of North Carolina at Chapel Hill, United StatesReviewed by:
Roberta Audrey Berard, Western University, Canada© 2023 Hetrick and Oliver. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Melissa Oliver bXNvbGl2ZXJAaXUuZWR1