 Cui-Jie Wei1,†
Cui-Jie Wei1,† Yi-Dan Liu1,†
Yi-Dan Liu1,† Yan-Ling Yang1
Yan-Ling Yang1 Yuan Wu1Jie-Yu Liu1
Yuan Wu1Jie-Yu Liu1 Xing-Zhi Chang1Ying Hua1
Xing-Zhi Chang1Ying Hua1 Yu-He Liu2
Yu-He Liu2 Hui Xiong1*
Hui Xiong1*
- 1Department of Pediatrics, Peking University First Hospital, Beijing, China
- 2Department of Otolaryngology Head and Neck Surgery, Capital Medical University Affiliated Beijing Friendship Hospital, Beijing, China
The TRNT1 gene encodes tRNA nucleotidyltransferase 1, which catalyzes the addition of cytosine-cytosine-adenosine (CCA) to the ends of cytoplasmic and mitochondrial tRNAs. The most common clinical phenotype associated with TRNT1 is autosomal recessive sideroblastic anemia with B-cell immunodeficiency, periodic fever, and developmental delay (SIFD). Muscle involvement has rarely been reported in TRNT1-related disorders. Here we report a Chinese patient with incomplete SIFD and hyperCKemia, and explored the skeletal muscle pathological changes. The patient was a 3-year-old boy with sensorineural hearing loss, sideroblastic anemia, and developmental delay since infancy. At the age of 11 months, significantly increased levels of creatine kinase were noted, accompanied by mild muscle weakness. Whole-exome sequencing revealed compound heterozygous variants of the TRNT1 gene, c.443C > T (p.Ala148Val) and c.692C > G (p.Ala231Gly), in the patient. Western blot showed a decreased expression of TRNT1 and cytochrome c oxidase subunit IV (COX IV) in the skeletal muscle of the patient. Electron microscopy observation of skeletal muscle pathology revealed abnormal mitochondria of various sizes and shapes, supporting a diagnosis of mitochondrial myopathy. The present case indicates that in addition to the classic SIFD phenotype, TRNT1 mutations can cause mitochondrial myopathy, a rare clinical phenotype of TRNT1-related disorders.
Introduction
The TRNT1 gene encodes tRNA nucleotidyltransferase 1, which is widely present in tissues in the whole body. The enzyme catalyzes the addition of cytosine-cytosine-adenosine (CCA) to the 3-prime end of tRNA precursors both in the cytoplasm and mitochondria (1–3). Clinical phenotypes associated with TRNT1 include autosomal recessive sideroblastic anemia with B-cell immunodeficiency, periodic fever, and developmental delay (SIFD, OMIM: 616084), and retinitis pigmentosa with erythrocytic microcytosis (OMIM: 616959). Till now, a total of 58 patients with SIFD and 41 mutations have been reported (4). SIFD is a autoinflammatory multisystem disorder, and the phenotypic spectrum is still emerging, with some specific common features but a diverse set of clinical phenotypes and patterns of system involvement described, thus it would be more appropriate to consider this syndrome as a TRNT1-related disorder (4). Muscle involvement has rarely been reported in TRNT1-related disorders. Only one patient has been reported to have a mild elevation of serum creatine kinase (CK) up to 383 U/L (5). Three other patients were found to have a decreased enzyme activity of the muscle mitochondrial respiratory chain, but none of them had clinical symptoms, such as muscle weakness or hyperCKemia (3, 5).
In this study, we report a Chinese child with TRNT1 gene mutations who had an incomplete SIFD phenotype and significantly elevated levels of CK. A skeletal muscle pathology examination was performed to explore the mechanism of muscle involvement.
Case description
The index case of this study was a 3-year-old boy who was admitted to the hospital due to sensorineural hearing loss, sideroblastic anemia, and developmental delay. He was born to non-consanguineous parents and was conceived by in vitro fertilization. The birth was uneventful at 37 + 3 weeks of gestation. He failed hearing screening after birth and was diagnosed with severe all-frequency sensorineural hearing loss at the age of 6 months, based on behavioral audiometry, auditory brainstem response (ABR), and distortion product otoacoustic emissions (DPOAE). After being fitted with a hearing aid, he responded to loud sounds. At the age of 8 months, routine examination showed mild microcytic hypochromic anemia with minimum hemoglobin of 88 g/L. Serum iron, ferritin, and serum transferrin saturation levels were normal. Iron staining of the bone marrow showed that ring sideroblasts accounted for 5%, supporting a diagnosis of sideroblastic anemia. During follow-up, the patient's hemoglobin level was 100–110 g/L. At the age of 11 months, hyperCKemia was noted in a routine examination. During follow-up, CK remained elevated, with a minimum of 724 U/L and a maximum of 13,280 U/L (concurrent serum myoglobin 1,106 ng/ml, reference value 0–70). Most of the time, the CK level fluctuated between 3,000–8,000 U/L (reference value 25–195). There was no relationship between CK value and exercise or infection. The patient was always weak and easily tired but had never experienced dark-colored urine. Psychomotor and physical development was abnormally delayed without seizures. He could sit unaided at 6 months and walk with support at 12 months but was never able to walk independently. He made little eye contact with others and still could not understand instructions or say any words. His growth also slowed down after the age of 1 year. His current weight and height of 12.7 kg and 90 cm, respectively, were less than the 3rd percentile of the same age and sex.
On admission, physical examination showed that the muscle strength was reduced in all limbs to grade 4 (based on the MRC scale), accompanied by significant hypotonia. Knee tendon and Achilles tendon reflexes were symmetrically elicited. After admission, laboratory examination revealed hemoglobin was 112 g/L. The immune function test showed that immunoglobulin G was reduced to 2.93 g/L (reference value 7–17), while peripheral blood T and B lymphocyte subsets were within the normal range. He had hyperlactatemia (3.0 mmol/L). The CK level was 3,321 U/L, while the serum myoglobin result was 330 ng/ml. Echocardiography showed ventricular septum hypertrophy. Visual evoked response and ABR testing could not elicit any waveform. Brain magnetic resonance imaging showed bilateral cerebral white matter hypomyelination. The Griffiths Mental Development Scale-Chinese was used to assess the development and showed a marked delay in the “Locomotor” (equivalent age of 11 months), “Personal-social” (equivalent age of 6.5 months), “Hearing and Language” (equivalent age of 5.5 months), “Hand-eye coordination” (equivalent age of 7.5 months) and “Performance” (equivalent age of 6 months) areas.
Trio-based whole-exome sequencing (Trio-WES) was performed (See the Supplementary Material for details). Candidate variants were validated by Sanger sequencing. Compound heterozygous variants were identified in TRNT1 (NM_182916.2), with c.443C > T derived from the mother and c.692C > G from the father (Figure 1). The pathogenicity of c.443C > T had been previously proved in a patient with SIFD (3). The variant c.692C > G was a novel variant and it was not present in the gnomAD and gnomAD_EAS databases. According to the American College of Medical Genetics and Genomics guidelines (6), the two variants could be considered “pathogenic” and “likely pathogenic”, respectively.
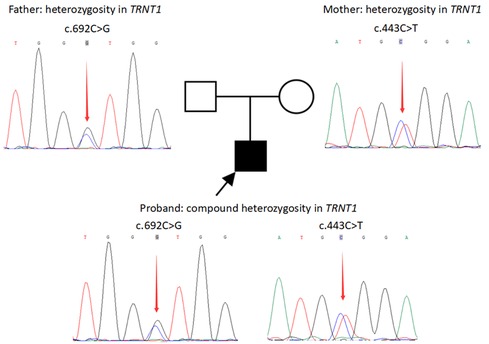
Figure 1. Variants in the TRNT1 gene in the family revealed by sanger sequencing. Compound heterozygous variants in TRNT1 (NM_182916.2) of the patient: c.443C > T derived from the mother and c.692C > G derived from the father.
PSIPRED V4.0 (http://bioinf.cs.ucl.ac.uk/psipred/) was further used to predict the secondary structures of the mutated and wild-type TRNT1 proteins. The variant c.443C > T (p.Ala148Val) was a missense variant that led to a change in the polarity of amino acids (Figure 2A). The other missense variant c.692C > G (p.Ala231Gly) resulted in changes in the secondary structures of the adjacent amino acids 226–227 and 232–233 from a coil to a helix (Figure 2B). The changes in local spatial structure were predicted by SWISS-MODEL (https://swissmodel.expasy.org/) (Figure 2C). A single nucleotide polymorphism (SNP) array, mitochondrial DNA sequencing, and trio-based whole-genome sequencing (trio-WGS) that were performed revealed no other variants associated with the phenotype of the patient (See the Supplementary Material for details).
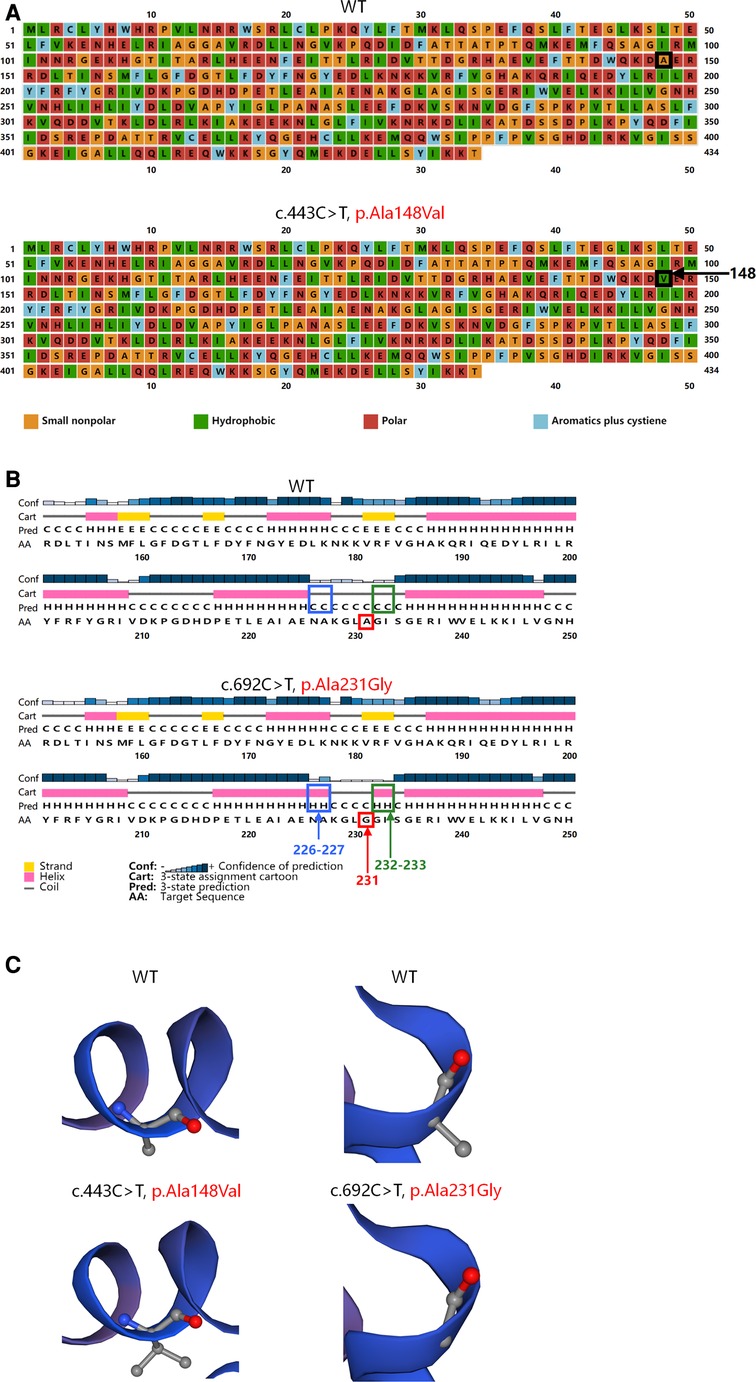
Figure 2. Structures of the wild-type and mutated TRNT1 proteins. (A) Variant c.443C > T (p.Ala148Val) led to a substitution of hydrophobic valine (green) for small nonpolar alanine (orange) at position 148 (black arrow) showed by PSIPRED. (B) Variant c.692C > G (p.Ala231Gly) (red arrow) resulted in changes in secondary structures of the adjacent amino acids 226–227 (blue arrow) and 232–233 (green arrow) from a coil to a helix predicted by PSIPRED. (C) Variant c.443C > T (p.Ala148Val) and variant c.692C > G (p.Ala231Gly) caused changes in local spatial structure of TRNT1 proteins predicted by SWISS-MODEL.
To clarify the cause of the high CK level, the right quadriceps muscle was biopsied. Light microscopy revealed slight pathological changes. Hematoxylin and eosin (HE) staining showed occasional necrotic muscle fibers. No typical “ragged-red” fibers (RRFs), “ragged-blue” fibers (RBFs), or cytochrome c oxidase (COX)-negative muscle fibers were found (Figure 3A). Electron microscopy showed that the mitochondria around the muscle fibers had different sizes and shapes. Some were empty or had discontinued ridges, supporting a diagnosis of mitochondrial myopathy (Figure 3B). Western blot showed a significantly decreased expression of TRNT1 in the skeletal muscle of the patient. A decreased expression of cytochrome c oxidase subunit IV (COX IV) protein was also detected (Figure 3C), indicating a damage of muscle mitochondrial respiratory chain complex IV (See the Supplementary Material for details of Western blot).
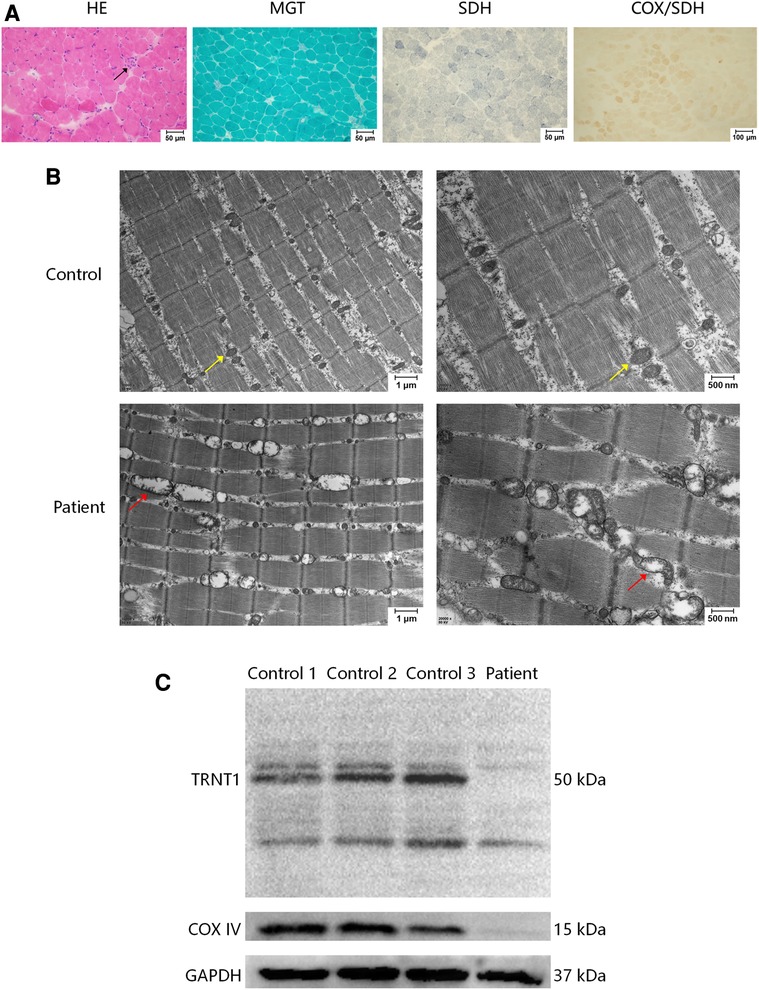
Figure 3. Muscle biopsy of right quadriceps muscle. (A) In a light microscopy, haematoxylin and eosin (HE) stain showed that polygonal muscle fibers were closely packed to each other, with occasional necrosis muscle fibers (arrow). No abnormities were detected in modified Gomori trichrome (MGT) staining, succinate dehydrogenase (SDH) staining, and combined cytochrome oxidase (COX)/SDH staining. (B) Electron microscopy showed that the mitochondria around the muscle fibers were of different sizes and shapes. Some were empty or had discontinued ridges (red arrows). The control showed normal mitochondria (yellow arrows). (C) Western blot of TRNT1, cytochrome c oxidase subunit IV (COX IV), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in human skeletal muscle samples of 3 independent controls and the patient. The patient showed a decreased expression of TRNT1 and COX IV. The primary antibodies are anti-TRNT1 polyclonal antibody (Abcam, ab224536), anti-COX IV monoclonal antibody (Gene-Protein Link, P01L08), and anti-GAPDH monoclonal antibody (Cell Signaling Technology, #2118).
Above all, according to Bernier et al. (2002) criteria for the diagnosis of mitochondrial respiratory chain disorders (7), the patient met the major clinical criteria for unexplained multisystem involvement, and other diseases were excluded by metabolic screening and genetic testing. He also met three minor diagnostic criteria: widespread electron microscopic mitochondrial abnormality in histology, a nuclear mutation of probable pathogenicity, and identification of hyperlactatemia in metabolism. Therefore, a definite diagnosis of mitochondrial disorders was established.
Discussion
TRNT1 is an essential enzyme that catalyzes the addition of the CCA trinucleotides terminus to the 3-prime end of tRNA precursors, which is necessary for aminoacylation (8). Mutations in the TRNT1 gene lead to an abnormal modification of cytoplasmic and mitochondrial tRNA, which further affects the formation of the mitochondrial respiratory chain complex, resulting in an oxidative phosphorylation defect and insufficient ATP supply. SIFD is the classic phenotype caused by TRNT1 gene mutations, which mostly involve the immune, blood, and nervous systems, but can also affect vision, hearing, heart, and kidneys (5, 9–12). However, only 65% of patients showed the complete phenotype characterized by anemia, recurrent fever, immunodeficiency and neurodevelopmental delay (4). In the present case, the child had an incomplete SIFD phenotype featuring low IgG, sideroblastic anemia, and developmental delay, but lacked the characteristic manifestation of periodic fever and B-cell immunodeficiency. Notably, this patient was accompanied by a significantly elevated CK, which has not been reported in the previous literature.
The human TRNT1 protein contains 434 amino acids and has two domains: a polymerase head domain (amino acids 59–182) and a probable RNA and SrmB-binding site of polymerase A (amino acids 215–271) (http://pfam.xfam.org/protein/Q96Q11). The variants c.443C > T (p.Ala148Val) and 692C > G (p.Ala231Gly) are located in the above two functional domains, respectively. And both variants are predicted to affect the spatial structure of the TRNT1 protein. Additionally, a decreased expression of TRNT1 protein has been observed in the skeletal muscle of the patient. SNP array, mitochondrial DNA sequencing, and trio-WGS found no other mutations that could explain myopathy. Therefore, we speculate that the hyperCKemia in this patient was likely attributed to TRNT1 mutations.
Skeletal muscles, as high energy-consuming tissues, are often involved in mitochondrial disease, and the associated clinical manifestations range from asymptomatic hyperCKemia, varying degrees of muscle weakness, to rhabdomyolysis induced by infection or exercise. However, there are few reports of muscle involvement in TRNT1-related disorders. Only one patient has been reported to have a mild elevation of CK to 383 U/L (5). Three other patients were found to have a decreased enzyme activity of muscle mitochondrial respiratory chain, but none of them had symptoms of myopathy, such as muscle weakness or hyperCKemia (3, 5). Unlike previous literature, the patients in this study presented with markedly elevated CK and mild muscle weakness. Moreover, his CK fluctuated greatly, with a maximum of 13,280 U/L, which was also in line with the characteristics of mitochondrial myopathy. The muscle pathology on light microscopy was relatively mild, and no RRFs or COX-negative muscle fibers were found. We speculate the reasons for the lack of RRFs or COX-negative muscle fibers: the first reason is that RRFs are rarely seen in patients with mitochondrial myopathy under 5 years of age; the second reason is that COX-negative muscle fibers are more common in mitochondrial gene mutations, but relatively rare for nuclear gene mutations. Electron microscopy observation of skeletal muscle pathology revealed abnormal mitochondria of various sizes and shapes, supporting a diagnosis of mitochondrial myopathy. Sasarman F et al. reported a patient harboring homozygous c.443C > T mutation in TRNT1 whose muscle autopsy showed enlarged mitochondria under electron microscopy (3), similar to our patient. Therefore, electron microscopy may be more helpful for discovering mitochondrial abnormalities in muscles in TRNT1-related disorders. Unfortunately, due to the limited amount of muscle samples of the patient, the enzyme activity of the muscle mitochondrial respiratory chain was not performed. But COX IV deficiency in the patient was detected by western blot, hinting a damage of muscle mitochondrial respiratory chain complex IV, whose activity was reduced in two previous reported patients with TRNT1 mutations (3, 5). Based on muscle weakness, significantly increased levels of CK, and the effect of TRNT1 mutations on muscle mitochondria, we diagnosed the patient with mitochondrial myopathy and gave him a “cocktail” medicine including levocarnitine, coenzyme Q10, and so on. After the treatment, his motor ability improved gradually, without developmental regression after infection.
In conclusion, this study describes a case with incomplete SIFD with prominent hyperCKemia caused by compound heterozygous variants in the TRNT1 gene, which indicates that in addition to the classic SIFD phenotype, TRNT1 mutations can cause mitochondrial myopathy.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author/s.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
C-JW and Y-DL: collected the data and wrote the paper. HX, C-JW, Y-LY, YW, YH, and Y-HL: participated in the diagnosis and treatment of the patient. C-JW, Y-DL, J-YL, and X-ZC: carried out the skeletal muscle pathology examination. HX: designed the present study and revised the paper. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the National High Level Hospital Clinical Research Funding (High Quality Clinical Research Project of Peking University First Hospital, No. 2022CR69), National Natural Science Foundation of China (No. 82171393), Natural Science Foundation of Beijing Municipality (No. 7212116), National Key Research and Development Program of China (No. 2016YFC0901505), Beijing Key Laboratory of Molecular Diagnosis and Study on Pediatric Genetic Diseases (No. BZ0317), and Peking University First Hospital Research Seed Fund (2019SF06).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2023.1160107/full#supplementary-material.
References
1. Wellner K, Betat H, Morl M. A tRNA's Fate is decided at its 3′ end: collaborative actions of CCA-adding enzyme and RNases involved in tRNA processing and degradation. Biochim Biophys Acta Gene Regul Mech. (2018) 1861:433–41. doi: 10.1016/j.bbagrm.2018.01.012
2. Mendonca LO, Prado AI, Costa IMC, Bandeira M, Dyer R, Barros SF, et al. Case report: expanding clinical, immunological and genetic findings in sideroblastic anemia with immunodeficiency, fevers and development delay (SIFD) syndrome. Front Immunol. (2021) 12:586320. doi: 10.3389/fimmu.2021.586320
3. Sasarman F, Thiffault I, Weraarpachai W, Salomon S, Maftei C, Gauthier J, et al. The 3′ addition of CCA to mitochondrial tRNASer(AGY) is specifically impaired in patients with mutations in the tRNA nucleotidyl transferase TRNT1. Hum Mol Genet. (2015) 24:2841–7. doi: 10.1093/hmg/ddv044
4. Maccora I, Ramanan AV, Wiseman D, Marrani E, Mastrolia MV, Simonini G. Clinical and therapeutic aspects of sideroblastic anaemia with B-cell immunodeficiency, periodic fever and developmental delay (SIFD) syndrome: a systematic review. J Clin Immunol. (2023) 43:1–30. doi: 10.1007/s10875-022-01343-0
5. Wedatilake Y, Niazi R, Fassone E, Powell CA, Pearce S, Plagnol V, et al. TRNT1 Deficiency: clinical, biochemical and molecular genetic features. Orphanet J Rare Dis. (2016) 11:90. doi: 10.1186/s13023-016-0477-0
6. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
7. Bernier FP, Boneh A, Dennett X, Chow CW, Cleary MA, Thorburn DR. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology. (2002) 59:1406–11. doi: 10.1212/01.wnl.0000033795.17156.00
8. Slade A, Kattini R, Campbell C, Holcik M. Diseases associated with defects in tRNA CCA addition. Int J Mol Sci. (2020) 21:3780. doi: 10.3390/ijms21113780
9. Hull S, Malik AN, Arno G, Mackay DS, Plagnol V, Michaelides M, et al. Expanding the phenotype of TRNT1-related immunodeficiency to include childhood cataract and inner retinal dysfunction. JAMA Ophthalmol. (2016) 134:1049–53. doi: 10.1001/jamaophthalmol.2015.5833
10. Frans G, Moens L, Schaballie H, Wuyts G, Liston A, Poesen K, et al. Homozygous N-terminal missense mutation in TRNT1 leads to progressive B-cell immunodeficiency in adulthood. J Allergy Clin Immunol. (2017) 139:360–3.e6. doi: 10.1016/j.jaci.2016.06.050
11. Lougaris V, Chou J, Baronio M, Gazzurelli L, Lorenzini T, Soresina A, et al. Novel biallelic TRNT1 mutations resulting in sideroblastic anemia, combined B and T cell defects, hypogammaglobulinemia, recurrent infections, hypertrophic cardiomyopathy and developmental delay. Clin Immunol. (2018) 188:20–2. doi: 10.1016/j.clim.2017.11.008
Keywords: TRNT1, mitochondrial myopathy, hyperCKemia, sensorineural hearing loss, sideroblastic anemia, developmental delay
Citation: Wei C-J, Liu Y-D, Yang Y-L, Wu Y, Liu J-Y, Chang X-Z, Hua Y, Liu Y-H and Xiong H (2023) Case report: Muscle involvement in a Chinese patient with TRNT1-related disorder. Front. Pediatr. 11:1160107. doi: 10.3389/fped.2023.1160107
Received: 6 February 2023; Accepted: 19 April 2023;
Published: 5 May 2023.
Edited by:
Magdalena Budisteanu, Prof. Dr. Alexandru Obregia Psychiatry Hospital, RomaniaReviewed by:
Duangrurdee Wattanasirichaigoon, Mahidol University, ThailandXin-Ming Shen, Mayo Clinic, United States
© 2023 Wei, Liu, Yang, Wu, Liu, Chang, Hua, Liu and Xiong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Xiong eGhfYmpiakAxNjMuY29t
†These authors have contributed equally to this work