 Rapeepat Thewamit1
Rapeepat Thewamit1 Chaiyos Khongkhatithum1
Chaiyos Khongkhatithum1 Lunliya Thampratankul1Wuttichart Kamolvisit2
Lunliya Thampratankul1Wuttichart Kamolvisit2 Arthaporn Khongkrapan3
Arthaporn Khongkrapan3 Duangrurdee Wattanasirichaigoon3*
Duangrurdee Wattanasirichaigoon3*
- 1Division of Neurology, Department of Pediatrics, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
- 2Division of Medical Genetics and Metabolism, Department of Pediatrics, King Chulalongkorn Memorial Hospital, Bangkok, Thailand
- 3Division of Medical Genetics, Department of Pediatrics, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Nonketotic hyperglycinemia (NKH) is in most cases a fatal inborn error of metabolism which usually presents during the neonatal period as encephalopathy and refractory seizures. The reported congenital anomalies associated with NKH included corpus callosal agenesis, club foot, cleft palate, and congenital heart disease. Here, we report a newborn who presented with encephalopathy without overt seizures, cerebral venous sinus thrombosis, and cleft palate. Electroencephalography showed a burst suppression pattern, which suggests the etiology could be due to a metabolic or genetic disorder. The amino acid analysis of plasma and cerebrospinal fluid showed elevated glycine. Whole exome sequencing identified a heterozygous c.492C > G; p.Tyr164Ter variant in exon 4 of the GLDC gene inherited from the patient's father. Further long-read whole genome sequencing revealed an exon 1–2 deletion in the GLDC gene inherited from the patient's mother. Additional analyses revealed no pathogenic variants of the cleft palate–related genes. The cleft palate may be an associated congenital anomaly in NKH. Regarding cerebral venous sinus thrombosis, we found a heterozygous variant (p.Arg189Trp) of the PROC gene, which is a common cause of thrombophilia among Thai newborns. A neonate with NKH could present with severe encephalopathy without seizures. A close follow up for clinical changes and further next generation sequencing are crucial for definite diagnosis in neonates with encephalopathy of unclear cause.
Introduction
Nonketotic hyperglycinemia (NKH) (MIM 605899) is a rare inborn error of metabolism (1). The disorder is caused by mutations of both alleles in the genes encoding proteins in the glycine cleavage system (GCS), which consists of glycine decarboxylase (P-protein: GLCD gene), aminomethyltransferase (T-protein: AMT gene), dihydrolipoamide dehydrogenase (L-protein: DLD gene), and glycine cleavage system H-protein (H-protein: GCSH gene). Patients with severe NKH often present with lethargy, coma, hypotonia, apnea, hiccups, and refractory seizures before 2 weeks of age. Moreover, patients have a high mortality rate (85%) (2, 3). Patients with attenuated NKH usually present after 2 weeks of age and have variable neurodevelopmental outcomes, including impaired verbal communication with/without epilepsy (3, 4). There have been reports of congenital anomalies, including cleft palate (4.6% or 3/65 cases), club foot, corpus callosal agenesis, and cardiac abnormalities in patients with NKH (2, 5), albeit with an unclear pathogenic mechanism.
Herein, we describe a newborn with NKH who presented with encephalopathy without clinical seizures, cerebral venosinus thrombosis, and concomitant cleft palate. Additionally, extensive genetic analysis, including second and third generation sequencing plus cytogenomic microarray, were performed in an attempt to identify the genetic defect underlying NKH, cleft palate, and thrombosis.
Case description
The patient was a 39-week gestational age male neonate, born via vaginal delivery following an unremarkable pregnancy with Apgar scores of 9 and 10 at 1 and 5 min, respectively, and a birth weight of 2,940 g. The patient was discharged to home on day of life (DOL) 2 with normal physical examination. At the age of 56 h (8 h after discharge), the patient was noted to become drowsy with a weak cry, necessitating an emergency visit and hospitalization at a local tertiary hospital. Physical examination revealed a temperature of 36.8°C, blood pressure of 75/30 mmHg, pulse rate of 140 beats/min, respiratory rate of 58 times/min, Glasgow Coma Score of 3 (E1V1M1), and pupils 2 mm, which were reactive to light. The muscle tone was hypotonic. Complete blood count, electrolytes, coagulogram (PT, PTT, fibrinogen level), arterial blood gases, and basic cerebrospinal fluid analysis were within normal limits. An amplitude-integrated electroencephalogram showed one suspicious electrographic seizure without a clinical seizure. Brain computer tomography (CT) showed minimal intraventricular hemorrhage at the bilateral occipital horn of lateral ventricles, thin subdural hemorrhage at the right tentorium, venous sinus thrombosis at the superior sagittal and transverse sinuses, and no congenital structural brain abnormalities. Phenobarbital was intravenously administered. The patient was intubated and transferred to our institution.
On DOL 3, at our hospital, the patient was comatose with apnea but without clinical seizure. Physical examination revealed cleft palate with no facial dysmorphic features, pupils 2 mm and normally reacting to light, the presence of doll eyes, generalized hypotonia, minimal spontaneous movement, deep tendon reflexes 1+, and absent Babinski signs. The Moro, suckling, and grasping reflexes were absent. The parents denied consanguineous marriage; however, they were from the same district. Both parents had no significant medical history of thrombosis. In addition, their first child had symptoms similar to the present patient and died on DOL 7 due to presumed infection.
The brain CT was reevaluated by our pediatric neuroradiologists, which showed cerebral venosinus thrombosis at the superior sagittal and transverse sinuses without evidence of cerebral edema or infarction (Figure 1). A brain MRI was not done. Protein C and protein S levels were not analyzed. Continuous electroencephalography (EEG) was performed, which showed a burst-suppression pattern without electrographic seizures (Figure 2). Pyridoxine 100 mg was intravenously administered and phenobarbital was continued; however, there was neither clinical nor EEG improvement. Given that the findings of brain CT could not explain the clinical findings of coma, we suspected that the etiology of coma and the burst suppression pattern on EEG could be an inborn error of metabolism or another rare genetic disease. Metabolic investigations were performed, which revealed a normal plasma ammonia level of 30 µmol/L (normal range 11–32 µmol/L), a mildly elevated lactate level of 2.5 µmol/L (normal range 0.5–2.2 µmol/L), and unremarkable urine organic acid profiles. Comprehensive metabolic screening (dry blood spot using tandem mass spectrometry) including acylcarnitine and amino acid profile analysis showed marked elevation of blood glycine (1,380 µmol/L; normal range 252–730 µmol/L). The CSF glycine level was 359 µmol/L (normal range 3.2–16.3 µmol/L). The patient's CSF/plasma glycine ratio was elevated at 0.31 (359/1,153 µmol/L). These results were consistent with severe NKH. Owing to the presence of cleft palate, cytogenomic microarray (CytoSNP-850 K, Illumina) was performed, which yielded no abnormality. Due to the severely poor prognosis of severe NKH, the parents were extensively counselled, and they decided to have the best supportive and symptomatic care for the patient. The family requested to have a peaceful terminal stage at their own place of residence. The patient passed away on DOL 7.

Figure 1. Contrasted brain CT of the patient. (A) Transverse sinus thrombosis. (B) Superior sagittal sinus thrombosis.
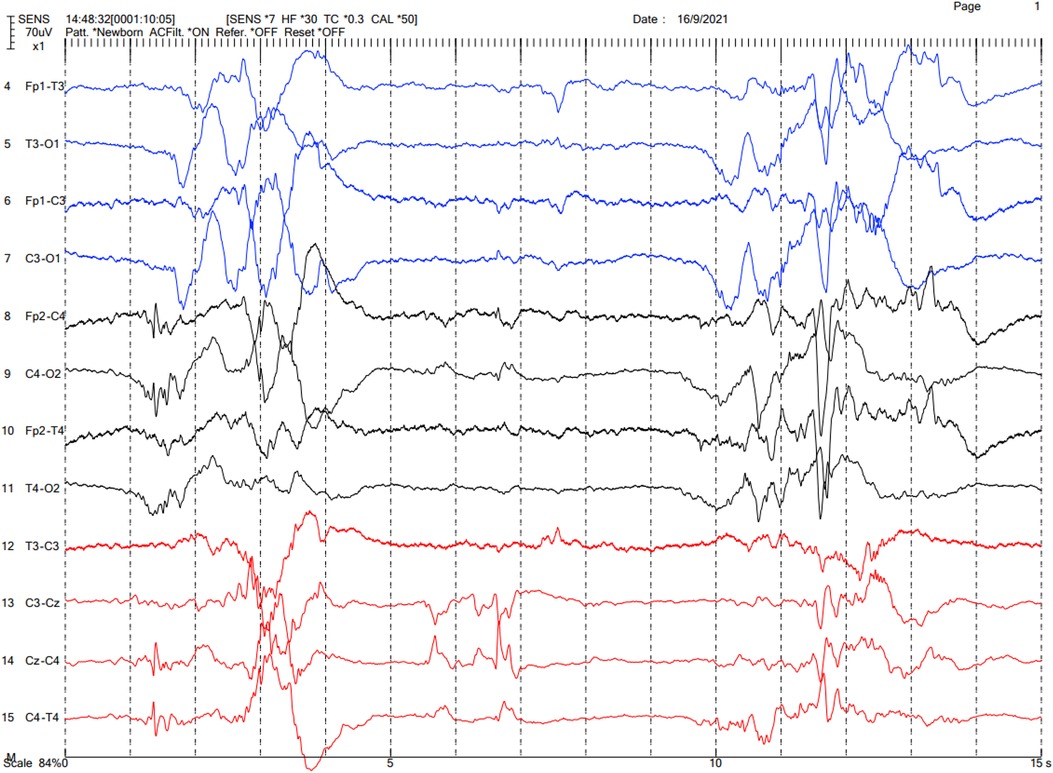
Figure 2. EEG of the patient showing the burst-suppression pattern.
Trio short-read whole exome sequencing (WES) with targeted gene analysis for hyperglycinemia (HP0002154: 19 genes) was performed, which revealed a heterozygous c.492C > G; p.Tyr164Ter (NM_000170.3) variant in exon 4 of the GLDC gene of the patient and the father, and no likely pathogenic/pathogenic variant in the GLDC gene of the mother (Figure 3A). Subsequently, long-read whole genome sequencing (WGS) was performed, which revealed exon 1–2 deletion in the GLDC gene of the patient and his mother. The methods for WES and WGS are provided in the Supplementary Material (6).
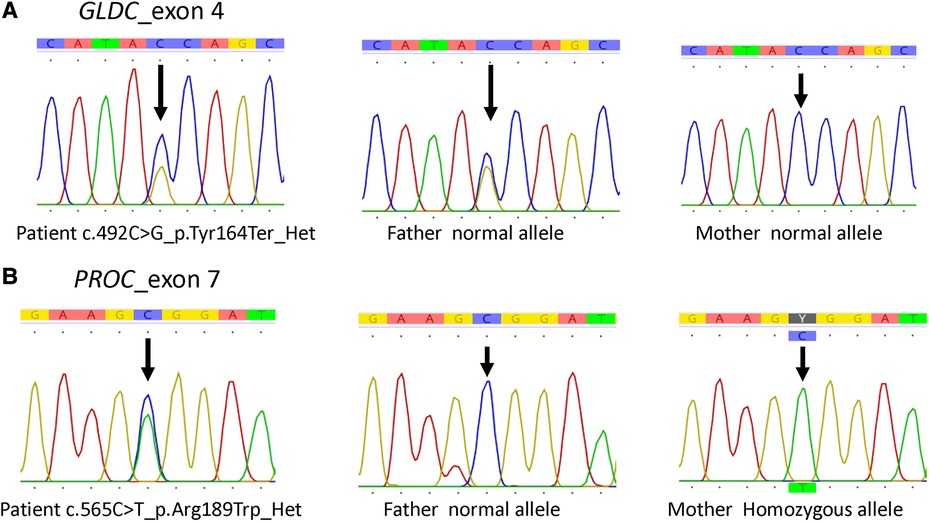
Figure 3. GLDC and PROC variants confirmed by Sanger sequencing of the family. (A) GLDC variants. (B) PROC variants.
Additionally, the human phenotype ontology (HPO) terms cleft palate (HP0000175: 548 genes) and venous thrombosis (HP0004936: 73 genes; plus PAI-1 gene) were analyzed to determine candidate variants underlying the cleft palate and cerebral venous thrombosis in the present patient. The analyses revealed no pathogenic/likely pathogenic variants of the cleft palate–related genes but a heterozygous pathogenic variant c.565C > T (p.Arg189Trp or Arg147Trp) in the protein C (PROC) gene (NM_000312.3) (Figure 3B). This PROC variant was inherited from his homozygous asymptomatic mother.
Discussion
We confirmed a neonate with glycine encephalopathy and cleft palate due to compound heterozygous pathogenic variants (a nonsense mutation in exon 4 and a deletion of exons 1–2) in the GLDC gene. The coincidental finding of venous thrombosis in the patient was likely associated with the PROC Arg147Trp variant identified.
According to a clinical survey of NKH, seizures, apnea, and hiccups were found in 85%, 79%, and 95% of patients with severe neonatal NKH, respectively, whereas the only consistent finding was hypotonia (100%) (7). The seizures in these cases usually began within the first few hours to days of life (2, 3). Glycine can be both an excitatory and inhibitory neurotransmitter. Glycine can stimulate N-methyl-D-aspartate (NMDA) receptors in the hippocampus, cerebral cortex, and cerebellum, causing seizures. Glycine also binds to glycine receptors in the brainstem and spinal cord, leading to apnea, hypotonia, and hiccups. Our patient presented with encephalopathy without overt seizures, but with venous sinus thrombosis and cleft palate, complicating the differential diagnosis. However, subsequent findings of a burst suppression pattern on EEG helped narrow down the diagnosis. The definite diagnosis was obtained from metabolic and molecular genetic tests. As a matter of fact, the patient might have had overlooked clinical seizure because it is difficult to observe the seizures of neonates.
Once NKH is suspected, the confirmation can be quickly achieved by measuring CSF and plasma glycine levels and the CSF/plasma glycine ratio, as shown in this case. The deletion of exons 1–2 was not detected by short-read WES but was picked up by long-read WGS. Recent data has indicated that the combination of long-read WGS and conventional WES provide a better diagnostic yield and may be a more economic approach in daily practice in the near future, given the rapid decline in cost (8). WGS covers exons, introns, and the intergenic sequence; therefore, it expands the analysis to regions not detectable by WES. WGS can detect balanced and unbalanced translocation, complex structural variants, and large deletion/insertion. It has been shown that WGS increased the incremental diagnostic yield by up to 10% in patients with intellectual disabilities and by up to 25% in children with other undiagnosed disorders, as compared to WES (9, 10).
Cleft lip/palate has been reported in 4.6% (3/65) of patients with NKH; however, detailed clinical findings and etiopathogenesis of these cases were lacking (2). The presence of cleft palate in our patient led us to hypothesize that the cleft might represent (1) a coincidental syndromic or nonsyndromic cleft or a common cleft palate due to multifactorial disorder in which no abnormality of a single gene/chromosome could be detected; or (2) a true association between the orofacial cleft and NKH, due to an uncharacterized mechanism.
Given the normal result of the microarray and the absence of cleft-related genetic variants in the present patient, chromosomal abnormality and single gene disorder as the cause of the cleft anomaly was likely excluded, leaving the possibility of multifactorial disorder, which cannot be confirmed by genetic testing.
The mechanism of the possible “true” association between NKH and the orofacial cleft has been mysterious (2, 7); though a direct or indirect teratogenic effect of intrauterine hyperglycinemia is possible. We searched the protein-protein interaction database STRING (https://string-db.org/) and found that SHMT2 (serine hydroxymethyltransferase), one among the top 10 proteins interacting with GLDC (Supplementary Material), was also listed as a cleft lip/palate–related gene, despite the absence of publication of SHMT2 mutations as a cause of orofacial cleft. SHMT2 plays a role in glycine and tetrahydrofolate metabolism in which the tetrahydrofolate pathway is known to be linked to isolated orofacial cleft (11). Whether or not SHMT2 is involved in hyperglycinemia-induced cleft palate requires further investigation.
Regarding cerebral venous sinus thrombosis, the risk factors associated with thromboembolism in neonates are central venous catheter, infection, asphyxia, polycythemia, and other predisposing factors. The heterozygous p.Arg189Trp variant of the PROC gene was likely the risk factor in the present patient. Protein C deficiency is a common risk factor of venous thromboembolism among Asian populations, including Thai, Chinese, and Japanese people (12–15). The disorder exhibits an autosomal dominant pattern with incomplete penetrance and variable expressivity (14). Homozygous individuals are more susceptible to develop venous thrombosis. The frequency of the Arg147Trp mutation in the PROC gene was prevalent in the Thai population; 9.5% for heterozygous and 2.7% for homozygous states (16). The Arg147Trp mutation increased the risk of cerebral venosinus thrombosis, with an odds ratio of 4.5 (95% CI: 1.6–12.8) for heterozygous and 43.3 (95% CI: 3.8–490.6) for homozygous states (16). A previous study found that both protein C and the functional level were decreased in Arg147Trp heterozygous individuals (14). An expression study of the PROC Arg147Trp variant in mammalian cell lines (HEK923) showed that the variant exhibited normal anticoagulant activity to Fva but a ∼3 times lower affinity for binding to endothelial protein C receptor (EPCR); thus the activation of this protein C variant on capillary endothelial cells could be potentially impaired and could contribute to the risk of developing venous thrombosis (14). Mutation of plasminogen activator inhibitor-1 (PAI-1), another thrombosis-related gene, was previously described in a neonate with NKH and cerebral venosinus thrombosis (17). The PAI-1 variant was not found in our patient. We, therefore, proposed that poor feeding, dehydration, and the presence of the PROC: p.Arg147Trp variant together contributed to the cerebral thrombosis observed in the present neonate.
Conclusion
We present a neonate with severe NKH in whom the presence of cerebral thrombosis together with cleft palate and initial absence of overt seizures complicated the diagnosis of NKH. A follow-up EEG looking for a burst suppression pattern and close monitoring for clinical changes are essential in neonates with encephalopathy of unclear cause, especially in countries lacking expanded newborn screening for inborn metabolic disorders.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Ethic Committee of Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
RT conducted data collection. RT and DW wrote the manuscript. DW, LT, and CK supervised this research. WK performed and interpreted the exome sequencing. AK performed molecular genetic testing. All authors performed critical reading and approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Acknowledgments
We thank the patient's family for their participation in this study, the Ramathibodi Foundation for financial support for the genomic testing, and the faculty of Medicine Ramathibodi Hospital for Research Career Development Award to DW. We kindly appreciate Prof. Vorasuk Shotelersuk, MD, Department of Pediatrics, King Chulalongkorn Memorial Hospital for exome sequencing support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2023.1155035/full#supplementary-material
References
1. Coughlin CR 2nd, Swanson MA, Kronquist K, Acquaviva C, Hutchin T, Rodriguez-Pombo P, et al. The genetic basis of classic nonketotic hyperglycinemia due to mutations in GLDC and AMT. Genet Med. (2017) 19(1):104–11. doi: 10.1038/gim.2016.74
2. Hoover-Fong JE, Shah S, Van Hove JLK, Applegarth D, Toone J, Hamosh A. Natural history of nonketotic hyperglycinemia in 65 patients. Neurology. (2004) 63(10):1847–53. doi: 10.1212/01.WNL.0000144270.83080.29
3. Van Hove JLK, Coughlin C II, Swanson M, Hennermann JB. Nonketotic hyperglycinemia. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al., editors. Genereviews((R)). Seattle, WA: University of Washington (2002). p. 1993–2023. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1357/
4. Swanson MA, Coughlin CR, Scharer GH, Szerlong HJ, Bjoraker KJ, Spector EB, et al. Biochemical and molecular predictors for prognosis in nonketotic hyperglycinemia. Ann Neurol. (2015) 78(4):606–18. doi: 10.1002/ana.24485
5. Al-Shareef I, Arabi M, Dabbagh O. Cardiac involvement in nonketotic hyperglycinemia. J Child Neurol. (2011) 26(8):970–3. doi: 10.1177/0883073811399150
6. Thongpradit S, Jinawath N, Javed A, Jensen LT, Chunsuwan I, Rojnueangnit K, et al. Novel SOX10 mutations in waardenburg syndrome: functional characterization and genotype-phenotype analysis. Front Genet. (2020) 11:589784. doi: 10.3389/fgene.2020.589784
7. Hennermann JB, Berger JM, Grieben U, Scharer G, Van Hove JL. Prediction of long-term outcome in glycine encephalopathy: a clinical survey. J Inherit Metab Dis. (2012) 35(2):253–61. doi: 10.1007/s10545-011-9398-1
8. Cohen ASA, Farrow EG, Abdelmoity AT, Alaimo JT, Amudhavalli SM, Anderson JT, et al. Genomic answers for children: dynamic analyses of >1000 pediatric rare disease genomes. Genet Med. (2022) 24(6):1336–48. doi: 10.1016/j.gim.2022.02.007
9. Bruel AL, Vitobello A, Tran Mau-Them F, Nambot S, Sorlin A, Denomme-Pichon AS, et al. Next-generation sequencing approaches and challenges in the diagnosis of developmental anomalies and intellectual disability. Clin Genet. (2020) 98(5):433–44. doi: 10.1111/cge.13764
10. Lionel AC, Costain G, Monfared N, Walker S, Reuter MS, Hosseini SM, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med. (2018) 20(4):435–43. doi: 10.1038/gim.2017.119
11. Garland MA, Reynolds K, Zhou CJ. Environmental mechanisms of orofacial clefts. Birth Defects Res. (2020) 112(19):1660–98. doi: 10.1002/bdr2.1830
12. Sirachainan N, Limrungsikul A, Chuansumrit A, Nuntnarumit P, Thampratankul L, Wangruangsathit S, et al. Incidences, risk factors and outcomes of neonatal thromboembolism. J Matern Fetal Neonatal Med. (2018) 31(3):347–51. doi: 10.1080/14767058.2017.1285892
13. Tsay W, Shen MC. R147w mutation of PROC gene is common in venous thrombotic patients in Taiwanese Chinese. Am J Hematol. (2004) 76(1):8–13. doi: 10.1002/ajh.20043
14. Ding Q, Yang L, Hassanian SM, Rezaie AR. Expression and functional characterisation of natural R147W and K150del variants of protein C in the Chinese population. Thromb Haemost. (2013) 109(4):614–24. doi: 10.1160/TH12-10-0760
15. Kinoshita S, Iida H, Inoue S, Watanabe K, Kurihara M, Wada Y, et al. Protein S and protein C gene mutations in Japanese deep vein thrombosis patients. Clin Biochem. (2005) 38(10):908–15. doi: 10.1016/j.clinbiochem.2005.05.006
16. Sirachainan N, Chuansumrit A, Sasanakul W, Yudhasompop N, Mahaklan L, Vaewpanich J, et al. R147w in PROC gene is a risk factor of thromboembolism in Thai children. Clin Appl Thromb Hemost. (2018) 24(2):263–7. doi: 10.1177/1076029617709085
Keywords: nonketotic hyperglycinemia, neonatal encephalopathy, cerebral venous sinus thrombosis, cleft palate, whole exome sequencing
Citation: Thewamit R, Khongkhatithum C, Thampratankul L, Kamolvisit W, Khongkrapan A and Wattanasirichaigoon D (2023) Case report: Severe nonketotic hyperglycinemia in a neonate without apparent seizures but concomitant cleft palate and cerebral sinovenous thrombosis. Front. Pediatr. 11:1155035. doi: 10.3389/fped.2023.1155035
Received: 31 January 2023; Accepted: 20 July 2023;
Published: 8 August 2023.
Edited by:
Madelyn Gillentine, Seattle Children’s Hospital, United StatesReviewed by:
Danijela Petković Ramadža, University Hospital Centre Zagreb, CroatiaDezhi Cao, Shenzhen Children’s Hospital, China
Francesca Furia, Filadelfia, Denmark
© 2023 Thewamit, Khongkhatithum, Thampratankul, Kamolvisit, Khongkrapan and Wattanasirichaigoon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Duangrurdee Wattanasirichaigoon ZHVhbmdydXJkZWUud2F0QG1haGlkb2wuYWMudGg=