 Abhay Katyal
Abhay Katyal Christopher O. Y. Li
Christopher O. Y. Li Sonia Franciosi
Sonia Franciosi Shubhayan Sanatani
Shubhayan Sanatani- British Columbia Children’s Hospital Heart Center, Department of Pediatrics, University of British Columbia, Vancouver, BC, Canada
Sudden cardiac death (SCD) is a rare and devastating event in children and remains a leading cause of death in young athletes. Channelopathies and cardiomyopathies, in particular long QT syndrome (LQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), hypertrophic cardiomyopathy (HCM), and arrhythmogenic cardiomyopathy (ACM) are associated with exercise-related SCD. Implantable cardioverter-defibrillators (ICDs) are often placed for secondary prevention for athletes with cardiomyopathy or channelopathy. There remains concern regarding the safety of return to participation with an ICD in place. Guidelines have historically recommended that patients with inherited heart rhythm disorders be restricted from competitive sports participation. Increasing evidence suggests a lower risk of exercise-related cardiac events in young athletes with inherited heart rhythm disorders. In this review, we highlight current knowledge, evolving guidelines, and present a multidisciplinary approach involving shared decision-making and appropriate planning for safe sports participation of children with inherited heart rhythm disorders.
Introduction
Sudden cardiac death (SCD) is a rare and devastating event in children and remains a leading cause of death in athletes aged 40 years or younger. Estimates show the incidence of SCD in young individuals (typically aged under 35–40 years) to be between 1 and 3 cases/100,000 (1). Exercise-related SCD is frequently caused by inherited heart rhythm conditions: channelopathies and cardiomyopathies. In particular long QT syndrome (LQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), hypertrophic cardiomyopathy (HCM), and arrhythmogenic cardiomyopathy (ACM) are associated with exertional ventricular arrhythmias. Guidelines recommend implantable cardioverter-defibrillators (ICDs) for secondary prevention in these conditions (2). Concern has been raised around sports participation for children with ICDs (3). Previous guidelines have recommended against sports participation for children with inherited heart rhythm disorders and ICDs due to an abundance of caution and fear of cardiac events (4).
Christian et al. reported that approximately half of their cohort of pediatric patients with either LQTS, CPVT, ACM, or HCM had reduced quality of life and modified their physical activity after receiving their diagnosis (5). Patients advised against physical activity because of their condition are at risk of gaining weight and obesity (5). Exercise restriction in children with inherited heart rhythm disorders can also lead to physical deconditioning and additional cardiovascular burden later in life (6). Reineck et al. found that the psychosocial and emotional well-being of HCM patients was negatively impacted due to exercise restriction (7).
Patients with inherited heart rhythm disorders are advised to modify their physical activity due to the perceived risk of exercise-related cardiac events. There is evidence that children with inherited heart rhythm disorders are at a lower risk of exercise-related cardiac events than previously thought (6, 8). Optimized medical management and creation of a safety plan are necessary to promote exercise participation for children affected by channelopathies and cardiomyopathies. The field is moving away from exercise restriction and towards shared decision-making, which places the patient at the center of the discussion (Figure 1). Appropriate risk assessment, regular expert follow-up, including a review of treatment compliance, increasing community awareness, and access to an automated external defibrillator (AED) are all important components to ensuring safe participation. In this review, we summarize current knowledge, guidelines, and present our multidisciplinary approach with appropriate planning in place so that pediatric patients with inherited heart rhythm disorders can participate in sports safely.
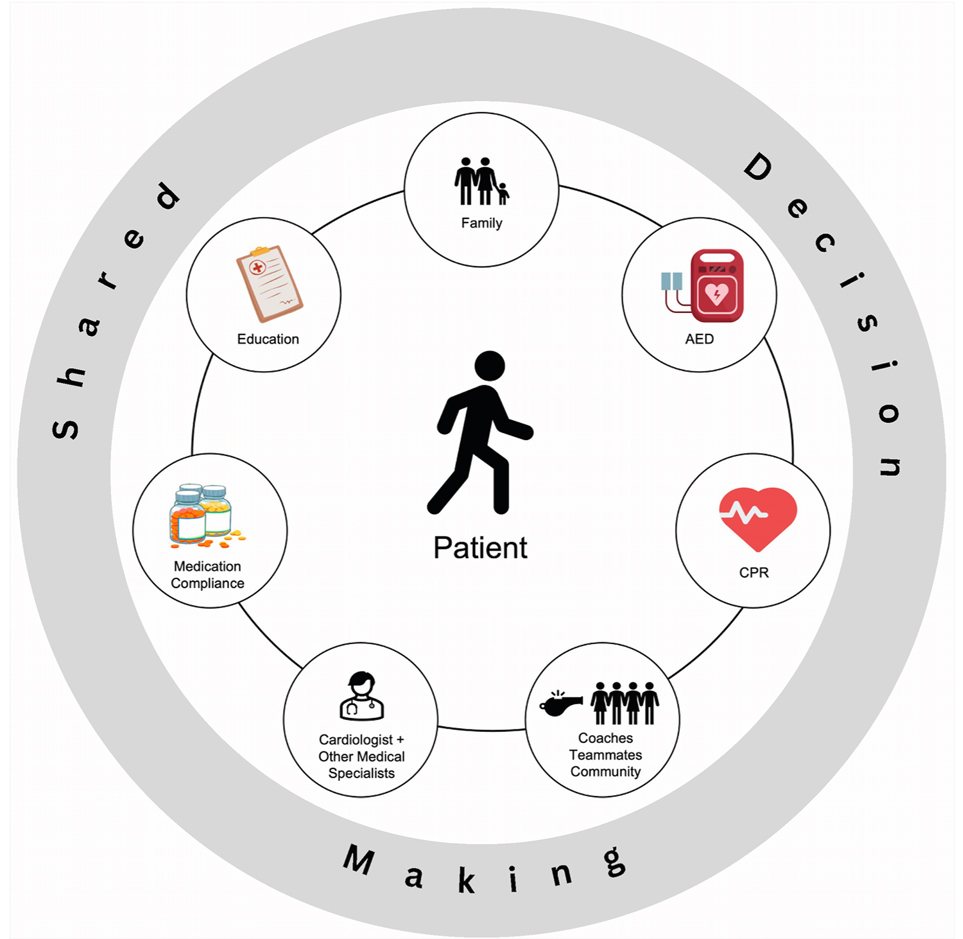
Figure 1. Shared decision-making model for sports participation. CPR, cardiopulmonary resuscitation; AED, automated external defibrillator.
Cardiac ion channelopathies & sports participation
Sports participation guidelines for channelopathies
In 2005, guidelines on exercise recommendations and sports participation were published by the European Society of Cardiology (ESC) and an expert task force from the 36th Bethesda Conference (4, 9). The American Heart Association (AHA), American College of Cardiology (ACC), and ESC subsequently published a report on the management of ventricular arrhythmias and prevention of SCD (10). These guidelines advised against competitive sports participation for inherited arrhythmias and were mostly based on expert consensus with low levels of evidence. Guidelines define a competitive athlete as an individual who participates in regular exercise training and competes in organized individual or team sports (9, 11, 12). This definition encompasses a wide range of ages as athletes can compete at various levels of competition (i.e., youth, high-school, college, national). Each guideline recommendation is categorized by a class of recommendation and level of evidence (LOE) (Table 1) (13, 14). Class I refers to strong recommendations where the benefits outweigh the risks. Class IIa (moderate) and IIb (weak) recommendations have some degree of benefit to the population. Class III reflects recommendations that either have no benefit or cause harm. LOE A is high quality evidence derived from multiple randomized-controlled trials or meta-analyses. LOE B is moderate quality evidence from other randomized or non-randomized studies that have not been externally validated. LOE C is given to guidelines based on either low quality of evidence or consensus of expert opinion. An international consensus statement published in 2013 from the Heart Rhythm Society (HRS), European Heart Rhythm Association (EHRA), and Asia Pacific Heart Rhythm Society (APHRS) on the diagnosis and management of patients with inherited primary arrhythmia syndromes followed a similar approach (15).

Table 1. Levels of evidence.
The 2015 AHA guidelines provided recommendations on sports participation for patients with cardiac ion channelopathies and presented new strategies for clinical management (16). Experts suggested that athletes should first be evaluated comprehensively by a heart rhythm specialist or genetic cardiologist (Class I; LOE C). If symptomatic, athletes should be restricted from participating in competitive sports until both they and their family are well informed, a treatment plan is in place, and the athlete has been asymptomatic after adhering to their therapy for more than three months (Class I; LOE C). They recommended that children can be considered for sports participation if they are asymptomatic or genotype-positive/phenotype-negative, and if certain precautionary measures are in place (e.g., having a personal AED and avoidance of dehydration/electrolyte imbalance) (Class IIa; LOE C). The 2020 and 2022 ESC guidelines remained stringent with their approach with few updates from their 2005 guidelines and recommended that symptomatic athletes be restricted from all competitive sports (12, 17). Specific recommendations for each channelopathy are outlined in each section below.
Long QT syndrome
Long QT syndrome (LQTS) is an inherited channelopathy characterized by a prolonged QT interval and T-wave abnormalities on the electrocardiogram (ECG). Approximately 1 in 2000 people are affected by LQTS, making it the most common cardiac channelopathy (18). The majority of patients have loss of function variants affecting the repolarizing potassium channels. The prolonged repolarization can lead to a characteristic type of ventricular tachycardia (VT) known as torsade de pointes, which causes syncope, sudden cardiac arrest (18), and SCD. Annual rates of syncope and SCD in untreated patients are 0.33%–0.9% and 5%, respectively (19). To date, mutations in 17 genes associated with LQTS have been found (20). The supporting data for many of these mutations is limited to small pedigrees and populations. The three most commonly implicated genes are KCNQ1, KCNH2, and SCN5A, and pathogenic variants in these three genes are responsible for 75%–80% of all congenital LQTS (21). β-blocker therapy remains the mainstay of treatment. Other interventions include avoidance of QT prolonging medications (www.crediblemeds.org), implantable cardioverter-defibrillators (ICDs) and left cardiac sympathetic denervation (LCSD).
In the past, children with LQTS had been advised against sports participation in accordance with guidelines and due to the perceived higher risk of recurrent cardiac events. Early data showed that exercise-related cardiac events in patients with LQTS were most commonly linked to the LQT1 genotype (62%), followed by LQT2 (13%) and LQT3 (13%) (18). LQT1 patients were observed to be at a higher risk of having cardiac events when exercising, especially during participation in activities involving high adrenergic stress (22). Swimming is a specific trigger in LQTS1(22). They observed that patients with LQT2 and LQT3 genotypes were more likely to experience cardiac events during rest/sleep (29% and 39%) compared to when exercising (13%). Auditory and emotional triggers of cardiac events were more prevalent among LQT2 patients (26%) compared to those with the LQT1 and LQT3 genotypes (2% and 7%).
What the guidelines say about LQTS
Previous guidelines recommended against competitive sports participation for LQTS patients (4, 9, 10). Guidelines from the 2005 36th Bethesda Conference advised that all patients with LQTS should be restricted from competitive sports, especially swimming, with the exception of minimal contact class 1A sports (e.g., billiards, bowling, cricket, curling, golf, and riflery) (4). In 2013, an HRS/EHRA/APHRS expert consensus statement modified their perspective for low-risk LQTS patients (15). Experts recommended that non-LQT1 genotyped asymptomatic patients with borderline QTc prolongation and no family history of SCD could be considered for competitive sports participation after a thorough clinical evaluation (Class I Recommendation). The 2015 AHA guidelines recommended that LQTS athletes who are symptomatic or have a baseline QT prolongation (QTc > 470 milliseconds in males and >480 milliseconds in females) could potentially participate in sports, except for competitive swimming in the LQT1 athlete (Class IIb; LOE C) (16). They further stated this would only be considered if they are asymptomatic on treatment for at least three months, not taking QT prolonging drugs, and follow appropriate precautionary measures. The 2020 ESC guidelines remained consistent with their previous recommendations in that high-intensity recreational and competitive sports participation should be avoided by LQTS patients, even when on β-blocker therapy (Class III; LOE B) (2, 12). Recent 2022 ESC guidelines recommended that LQTS patients avoid QT-prolonging drugs, avoid and correct electrolyte abnormalities, and avoid genotype- specific triggers for arrhythmias as described above (17).
Current LQTS sports participation evidence
Evidence supporting return to play for children with LQTS has grown significantly. In 2013, Johnson and Ackerman determined the outcomes of LQT1–3 patients who chose to continue to participate in competitive sports despite guideline recommendations (23). From a cohort of 353 patients, 130 (37%) participated in competitive sports, including 20 with ICDs. There were no LQT-triggered cardiac events among competitive athletes in over 650 patient-years. Medication compliance was implicated in two breakthrough cardiac events that occurred while preparing to compete in sporting events. In 2015, Aziz et al. conducted a retrospective review of genotype-positive LQTS pediatric patients to determine prevalence and outcomes of sports participation (24). Of the 212 patients who received β-blocker therapy, 26 (26%) participated in competitive sports and 77 (75%) participated in recreational sports. There was non-compliance and intolerance to β-blockade treatment in two patients. No patients experienced LQTS symptoms or cardiac events while participating in sports. These results highlight the importance of medication compliance.
Recent data further supports the safety of sports participation in LQTS patients. Chambers et al. reported no cardiac events or deaths in 106 athletes who participated in competitive sports (25). Four patients who were previously symptomatic with very prolonged QT intervals had cardiac events during recreational exercise. Tobert et al. reviewed the electronic medical records of 494 LQTS patients and reported no exercise-related deaths with 2056 combined years of follow-up (21). Three athletes (0.6%) experienced a cardiac event related to sports participation. In 2022, our center reported that physical activity levels were similar in 23 LQTS pediatric patients compared to healthy controls (26). These studies also support the safety of a shared decision-making model.
Catecholaminergic polymorphic ventricular tachycardia
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a primary electrical disease characterized by bidirectional and polymorphic VT during adrenergic stress, normal resting ECG, and a structurally normal heart (27). It is a rare condition estimated to affect 1 in 10,000 individuals (15). An individual with CPVT may experience palpitations, light-headedness, syncope, and sudden cardiac arrest (SCA)/SCD during physical activity or emotional stress. CPVT is caused by abnormal calcium handling in the cardiomyocyte. This is most commonly caused by gain-of-function variants in the cardiac ryanodine receptor-2 (RYR2) gene and is inherited in an autosomal dominant manner. CPVT can also be caused by genetic variants in the genes encoding calsequestrin-2 (CASQ2) and calmodulin (CALM1) (28).
The current treatment strategies for CPVT include β-blockers as the first-line treatment, with flecainide, LCSD, and ICDs as additional therapies (29). Flecainide has been increasingly used in treating CPVT in the past decade (30, 31). The correct medication and dosage, along with ensuring medication compliance is important in managing CPVT.
What the guidelines say about CPVT
The 2005 36th Bethesda Conference guidelines recommended that all patients with CPVT should be restricted from competitive sports, especially swimming, with the exception of minimal contact class 1A sports (4). Experts indicated that a less restrictive approach may be considered for genotype-positive/phenotype-negative athletes. The 2013 HRS/EHRA/APHRS international consensus statement and 2015 ESC guidelines recommended that CPVT patients limit or avoid competitive sports, strenuous exercise, and stressful environments (Class I; LOE C) (2, 15). The 2015 AHA guidelines suggested against competitive sports participation for previously symptomatic or asymptomatic CPVT athletes if they have exercise-induced premature ventricular contractions (PVCs) in bigeminy, couplets, or non-sustained VT (Class III; LOE C) (2, 16). Although the 2015 AHA guidelines advised disqualification, they stated that the athlete/family could potentially consider sports participation after a thorough consultation with a CPVT specialist (16). The 2022 ESC guidelines recommended that patients with CPVT should avoid competitive sports, strenuous exercise, and stressful environments (Class I; LOE C) (17).
Current CPVT sports participation evidence
Current evidence challenges exercise restriction in CPVT patients. The risk of recurrent cardiac events is lower than reported in early series, especially during exercise (32, 33). In fact, exercise may be beneficial in CPVT. In 2012, Kurtzwald-Josefson et al. found that exercise training can improve cardiac function and reduce arrhythmia in CASQ2 knockout mice that have a similar phenotype to human CPVT2 (34). Faster supraventricular rates induced with atropine can prevent ventricular arrhythmias, bidirectional VT, and ventricular ectopic beats during exercise in CPVT mice and patients (35, 36).
Ostby et al. conducted a retrospective study to investigate the outcomes in CPVT patients who were non-athletes (n = 43) or continued to participate in sports (n = 21) (37). Eight of 21 (38%) patients competed competitively as college athletes or adults. Only three athletes (14%) had cardiac breakthrough events: two were associated with recreational exercise and one was due to non-compliance with β-blocker therapy. Six non-athletes (14%) who were symptomatic had CPVT-linked cardiac events, and two non-athletes did not comply with their β-blocker treatment. This further underscores the importance of medication compliance in preventing events.
Cardiomyopathies & sports participation
Sports participation guidelines for cardiomyopathies
Due to the risk of SCA/SCD during sports and exercise particularly in young patients with cardiomyopathies, guidelines were developed to reduce risk through disease specific management and activity restriction. Previous guidelines advised routine disqualification of athletes diagnosed with cardiomyopathies from competitive sports (9, 38). ACC/AHA & European Association of Preventive Cardiology (EAPC) guidelines have shifted to a more case-by-case basis with a shared decision-making approach involving the patient, family members, and physician/cardiologists (12, 39, 40). Coaches should be informed of the patient's condition to ensure ongoing surveillance including potential occurrence of SCA during sports participation as well as to ensure that an emergency action is in place if a SCA were to occur (38). Return to play has not been well studied in young competitive athletes with cardiomyopathies.
Arrhythmogenic cardiomyopathy
ACM is an inherited structural heart disorder that predisposes individuals to ventricular arrhythmias and SCA. It typically presents between the second and fourth decade of life. In ACM, risk of arrhythmias and disease progression is accelerated by exercise (41). It presents more often as SCA/SCD in pediatric patients than in adults (42, 43). Structural changes are characterized by an enlarged, dilated right/left ventricle with or without decreased systolic function. There is fibro-fatty degeneration of the right ventricular (RV) (5) and/or left ventricular (LV) (44) myocardium which is associated with frequent arrhythmias. Most cases are inherited in an autosomal dominant manner, and due to causative variants in genes encoding the desmosome. During early stages of the disease, there is a subclinical phase where structural abnormalities are concealed and patients exhibit no symptoms. Patients are still at risk of experiencing SCA/SCD during this phase (45). This concealed phase then progresses to an electrical phase characterized by abnormalities on ECG including T-wave inversions, PVCs, and VT with left bundle branch block morphology. The structural phase presents with phenotypic alterations including right or biventricular dilatation and potentially heart failure.
Task force criteria may underestimate incidence of ACM in children (46). The ECG is insensitive in detecting ACM in young asymptomatic patients (47). Characteristics on ECG, PVC burden, complex PVC, cardiac magnetic resonance (CMR) results are important for diagnosis and determining disease progression (43, 45). Further family history, genetic testing, multimodal imaging and reassessment as the child grows are crucial in order to determine diagnosis and treatment (48). A risk model for ACM encompassing age, sex, syncope, nonsustained VT, PVCs, T-wave inversions on ECG, and RV ejection fraction has been developed for adult patients, but this has not been validated yet in pediatric patients (13).
What the guidelines say about ACM
The 2005 ESC guidelines recommended against participation in competitive sports for ACM patients (9). An ACC/AHA 2015 expert task force advised that ACM patients should not participate in most competitive sports, with the possible exception of class 1A sports (Class III; LOE C) (38). In 2015, ESC experts recommended avoidance of competitive sports, remaining stringent with their approach as there were no controlled trials showing beneficial effects of exercise in ACM patients (Class I; LOE C) (2). Since 2017, both national and international guidelines have suggested that ACM patients should avoid competitive or high-intensity endurance exercise as they are linked to increased risk of ventricular arrhythmias and disease progression (12, 49, 50). The 2020 ESC guidelines recommended that ACM patients should participate in 150-minute low intensity exercise per week (Class IIa; LOE C) (12). Experts stated that participation in low-to-moderate intensity recreational exercise/sports may be considered in patients with no history of cardiac arrest and minimal structural abnormalities (Class IIb; LOE C). The 2022 ESC guidelines recommended against participation in high-intensity/competitive sports for ACM patients including those who are genotype-positive/phenotype-negative (Class IIb, LOE C) (17). Gow et al. demonstrated that children with inherited arrhythmia conditions routinely exceed recommended activity levels (51). Evidence of disease progression and worse outcomes in 80 subjects with the TMEM43 p.S358L ACM mutation who participated in high-intensity exercise [>9 metabolic equivalent of task (MET)-hours/day] led to the recommendation that cascade screening be done earlier, to inform exercise choices reflective of the genotype (52). Cascade screening is a systematic approach of identifying family members of a proband who may be at risk for an inherited arrhythmia condition via clinical screening and genetic testing (53).
Current ACM sports participation evidence
Exposure to exercise accelerates ACM disease progression and reducing exercise can lower the risk of experiencing ventricular arrhythmias (41). Pediatric-onset ACM has been associated with increased endurance activity compared to adult-onset disease (42). Lie et al. showed that adult ACM patients were more likely to experience ventricular arrhythmias if they participated in high-intensity exercise (74%) and long-duration exercise (65%) (54). Phenotype-negative desmosomal gene-positive females who participated in high-intensity and long-duration exercise during adolescence were at greater risk of developing ACM (55). Wang et al. found that ACM patients with primary prevention ICDs and those who were gene-elusive benefited from exercise reduction (56). Adult TMEM43 positive ACM patients who participated in high-intensity exercise in the year prior to ICD implantation experienced a 9-fold-increase in appropriate ICD discharge compared to patients who participated in moderate-intensity exercise (52). Half of previously athletic ACM patients still experienced ventricular arrhythmias despite exercise reduction, therefore suggesting that ICDs should continue to be recommended if high-risk features exist (56). Smith et al. found that desmoplakin associated cardiomyopathy showed no correlation between exercise and risk of ventricular arrhythmias or LV/RV dysfunction, indicating that risk of arrhythmias may be specific to genotype (57). Risk and outcomes in ACM can change over time, therefore, it is imperative that ACM patients engaged in low or moderate activity be assessed yearly (12, 58).
Adolescents who are genotype-positive/phenotype-negative should be potentially restricted from high intensity sports and counselled about possible long term risks (48). Activities such as soccer, aerobics, or fast competitive swimming on a regular basis should be avoided. Restriction from competitive sports participation is regarded as an important preventative tool for both asymptomatic (gene-positive) and symptomatic patients as it can slow disease progression and prevent life threatening arrhythmias (48). To date, there has been no compelling data supporting the safety of exercise in ACM; in particular, data examining pediatric patients is lacking.
Hypertrophic cardiomyopathy
HCM is characterized by an abnormally thickened ventricular myocardium that can manifest at any age (59). There are a number of causes, particularly in infants and children, in whom inborn errors of metabolism and syndromic causes predominate (60). In adults, the disease is caused by variants in genes encoding the thick and thin contractile myofilament protein components of the sarcomere or adjacent Z discs. Inheritance is autosomal dominant with variable penetrance. The HCM phenotype varies from asymptomatic to LV outflow tract obstruction, diastolic dysfunction, heart failure, tachyarrhythmias and SCA/SCD. Although the presence of LV hypertrophy is considered a diagnostic hallmark of the disease, arrhythmia, SCA/SCD may present prior to structural changes (61). Clinical evaluation should include family history, echocardiogram, ECG, Holter monitoring and exercise stress test to determine risk for sudden death. CMR with late gadolinium imaging should be considered in addition to echocardiography to determine the scar burden (62). The phenotype can progress as the child grows. Models for SCD risk prediction in pediatric HCM have been developed (63, 64). The risk models include predictors such as age, maximal LV wall thickness, left atrial diameter, LV outflow tract gradient, family history of SCD, non-sustained VT, and unexplained syncope. LV outflow tract obstruction can increase during exercise and ischemia may result. HCM pathology is associated with myocyte disarray and ischemia. In the young, the extent of HCM pathology was found to be greater than older patients and correlated with an abnormal vascular response to exercise (65).
What the guidelines say about HCM
The 2005 ESC guidelines stated that physical activity could induce hypertrophy and recommended against competitive sports participation for HCM patients (9). Guidelines from the 36th Bethesda conference indicated that HCM athletes may be at a greater risk for SCA/SCD during exercise (4). The 2014 ESC guidelines on the diagnosis and management of HCM advised against participation in competitive sports and intense physical activity (66). The 2015 ACC/AHA guidelines restricted patients with a probable HCM diagnosis from participating in competitive sports, with the possible exception of class 1A sports (Class III; LOE C) (38). Experts stated competitive sports participation may be considered for asymptomatic genotype-positive HCM patients without evidence of LV hypertrophy, especially if they have no family history of HCM-related sudden death (Class IIa; LOE C). More recent 2020 ACC/AHA and ESC guidelines have shifted to the perspective that high-intensity recreational activities or moderate-to high-intensity competitive sports may be considered after a comprehensive evaluation and shared decision-making approach (12, 39, 40).
Current HCM sports participation evidence
HCM is a frequent cause of SCD in young competitive athletes (67). In a HCM mouse model, exercise before development of the phenotype prevented myocyte fibrosis and other markers of myocardial hypertrophy. Exercise reversed myocyte disarray and other markers of hypertrophy in older mice (68). In 1998, Corrado et al. reported 49 sudden deaths within a cohort of 33,735 competitive athletes (mean age of 19 years) who underwent pre-participation screening (69). 22 athletes who had definite evidence of HCM were disqualified from sports participation and no mortality was reported during follow-up. This data indicated that pre-participation screening in detecting HCM can be beneficial in reducing mortality. Malhotra et al. screened 11,168 adolescent soccer players (mean age of 16 years) and 5 were identified to have HCM (70). Over a follow-up period of 10.6 years, 3 of the 5 patients stopped to play and survived whereas two players continued to play and died.
Training early in young HCM patients may be beneficial in preserving LV diastolic function later in life (71). Greater exercise training during childhood and adolescence in HCM phenotype-positive or genotype-positive patients correlated with more favorable LV relaxation, filling, and end diastolic LV volume (72). Young HCM patients who participated in competitive sports have also been found to have a larger LV cavity size and better diastolic function compared to sedentary HCM patients (73). Exercise can reduce blood pressure (74). As hypertension accelerates HCM and is known to be a major cause of LV hypertrophy, exercise could be beneficial in limiting adverse hypertensive effects (75–77). Among 31 low-risk athletes with HCM, there was no difference in incidence of symptoms or major cardiac events between athletes who stopped exercise and those who continued to participate in competitive sports (78). Athletes were classified as either high-risk or low-risk according to risk factor profile scores set by SCD algorithms developed by the AHA and ESC (79). An HCM athlete is considered as low-risk if they have less than four of the following risk factors for SCD: non-sustained VT, severe LV hypertrophy, family history of SCD, unexplained syncope, and abnormal blood pressure response to exercise. Pelliccia et al. conducted a follow-up study with low-risk HCM phenotype patients (19–44 years old) who either continued to participate in competitive sports or reduced or stopped exercise (80). Over a follow-up period of 7 years, there was no difference in risk of SCA/SCD between HCM-trained and detrained patients. Therefore, in low-risk HCM patients, participation in competitive sports does not appear to increase risk of cardiac arrest or death. These results may not reflect those with a more severe HCM phenotype or younger adolescent HCM patients who are greater risk of SCD.
Safety of sports for young patients with ICDS
ICDs are recommended for most patients following cardiac arrest for secondary prevention of SCA. In some cases, ICDs for primary prevention are implanted in those considered at high arrhythmic risk. The risk-benefit balance of primary prevention ICDs in the pediatric population are not well established. However, ICDs in young patients with inherited heart rhythm disorders are associated with a longer-term risk of inappropriate shocks and device-related complications (81). While life-saving, adjustment to life with an ICD can be challenging (82). Risk factors associated with poor coping after ICD implantation include young age and frequent shocks (83).
What the guidelines say about ICDs
Prior to 2015, experts recommended against competitive sports participation for children with ICDs due to higher perceived risks as there could be potential failure of the ICD to defibrillate, risk of death due to ICD shock, or damage to the device. After initial supporting results from the ICD Sports Safety Registry, the 2015 ACC/AHA guidelines stated that competitive sports participation could be considered for athletes with ICDs (16, 84). Expert consensus indicates that the decision whether a young patient with an ICD participates in sports should be patient specific (44). The underlying cardiac disease, type of device, age, and activity needs to be considered. ICD implantation is not advised in asymptomatic patients with a diagnosis of LQTS or CPVT (Class III; LOE C). If β-blockade treatment is ineffective or intolerable for symptomatic LQTS patients and other interventions (e.g., LCSD) are not effective, then ICDs are recommended (Class I; LOE B). ICDs are indicated for symptomatic CPVT patients who have experienced cardiac arrest or refractory ventricular arrhythmias despite being on maximally tolerated combination medical therapy (Class I; LOE C). ICDs are recommended for ACM patients with sustained VT, syncope associated to ventricular arrhythmia, or an LV ejection fraction ≤35% (Class I; LOE B). In HCM patients with primary risk factors (e.g., LV hypertrophy, unexplained syncope), ICDs are recommended after considering the complications that can arise given the disease progression.
ICDs should not be implanted for the purpose of sports participation (Class III; LOE B) (44). Once an athlete with an ICD returns to play, medications and device settings should be optimized. Programming the ICD with high-rate cut-offs and long-detection duration in athletes can reduce inappropriate shocks without affecting survival (85). Further, an emergency action plan, as we discuss in further detail below, consisting of informed and cardiopulmonary resuscitation (CPR) trained coaches and staff, quick action plans, and availability of an AED is crucial for safe return to play (86).
ICDs and current sports participation evidence
The multinational ICD Sports Safety Registry was established to assess the risk of sports participation for athletes with ICDs (84). 328 athletes (10–60 years old) with ICDs participating in competitive sports were recruited. Results showed that there were no occurrences of death, resuscitated arrhythmia, or shock-related injury during sports participation (84, 87). Saarel et al. conducted a subanalysis of the registry and collected data on sports and clinical outcomes from a cohort of 129 young athletes (10–21 years old) (3). These athletes were followed over a median of 3.5 years, and the most common diagnoses were LQTS, HCM, and congenital heart disease. Results indicated that young patients with ICDs can participate in competitive and high-intensity sports without failure of ICDs to terminate arrhythmias or injury (3). Sports participation was not associated with significant adverse events, although appropriate and inappropriate shocks did occur (3, 84, 87).
Multidisciplinary approach & safe sports participation recommendations
It is important to discuss activity participation upon making the diagnosis of an inherited heart rhythm condition. There are a lot of misconceptions, and the tendency is to restrict activity without necessarily enabling participation. It is important that patients understand what their diagnosis is, and the specifics of that condition. Over the past three decades, causal genes implicated in inherited heart rhythm disorders have been successfully identified, and this has played a significant role in how these diseases are diagnosed and managed today. This means that pre-symptomatic patients and those without a phenotype are being diagnosed. The importance of genetic counselling in probands with inherited arrhythmias and family members has become widely recognized. Confronting patients with the risk of SCD and the possibility of transmitting the potentially fatal disease to their relatives may result in adverse psychological consequences; this distress highlights the importance of psychological support for these patients (88).
The patient and physician should engage in shared decision-making when discussing exercise participation (Figure 1). Shared decision-making considers patient goals and preferences balanced against the risk and benefits of exercise using clear communication. This discussion should include the most current evidence, nature of the athletic activity, sport-specific risks, and individual athlete characteristics and goals, all to arrive at patient-centered treatment decisions. It is important to bear in mind that current exercise guidelines are not exclusions in absolute terms. Almost all patients with inherited heart rhythm conditions can and should participate in exercise. It is important that patients find the most suitable exercise for their condition and lifestyle. Our starting point is that there are no exercises or sports that are not open for discussion. Rather, the focus is encouraging the patient to participate if they can develop a feasible safety plan for the activity. It is important to promote activity, not just remove restriction. The exception to this approach may be ACM, in which prolonged and intense exercise may accelerate the disease progression. It is also important to emphasize that prolonged and highly intense exercises are not required for patients to obtain cardiovascular benefits.
Our knowledge and the patient's condition can change over time, and therefore patients should attend regular appointments with their healthcare providers. Those choosing to participate in exercise should be made aware of benign and potentially serious cardiac symptoms that may occur during activity. Patients should rest if they notice these symptoms and contact their healthcare provider for further evaluation (Figure 2). Medication compliance is of utmost importance. Strenuous activity in hot environments, dehydration, and poor nutrition preceding activity can lead to electrolyte disturbances; adequate hydration and nutrition are always advised prior to exercise participation.
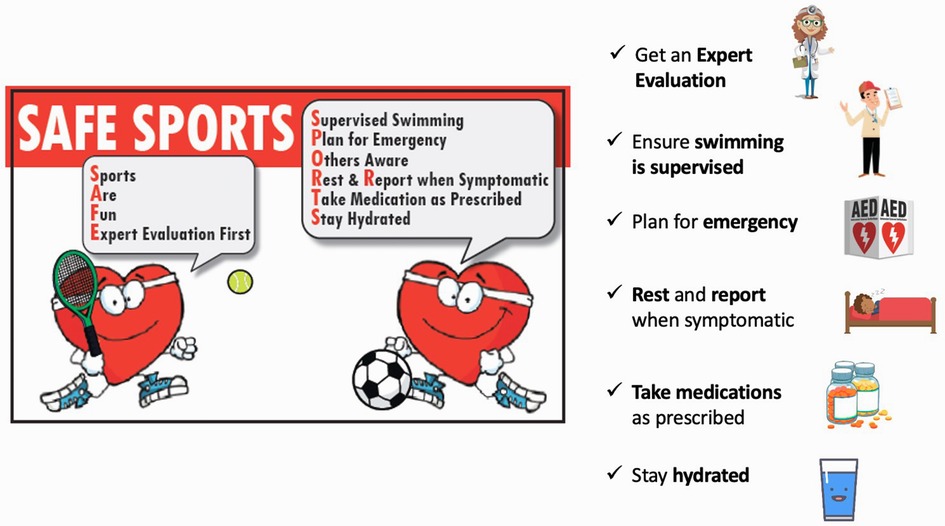
Figure 2. Safety of sports in children with inherited heart rhythm disorders. Modified from Cheung et al., 2016 (89).
The inherited heart rhythm clinic should support the athlete by providing clear communication to families, coaches, trainers, and schools. Athletes and those around them including family, teammates, coaches, trainers, and school staff should have a plan in place in the event of a potential SCA. This includes having protocols in place to notify first responders, knowing where to retrieve the AED, and educating personnel in CPR and AED use (90). Team sports or exercises with peers are preferred in the event that help is required. Coaches and schools can incorporate AEDs into team equipment. AEDs should be available and rapidly accessible at home, in the community, and at competitions. Training for this life-saving skill is easy to access through many different local organizations.
Conclusions
Sports participation in children with inherited heart rhythm disorders has been widely debated by experts in the field, with a tendency to restrict activities extensively. Exercise restriction can have a negative impact on the child's overall quality of life and well-being. Current data where available have not supported the benefit of such restrictions. Activity can be safely promoted in all conditions with perhaps the exception of ACM. A multidisciplinary approach that includes shared decision-making with appropriate risk assessment and follow-up, and development of safety plans can lead to healthier lifestyles. Increased community awareness regarding CPR and AED use are important measures for the rare breakthrough events.
Author contributions
All authors contributed and are responsible for drafting and reviewing the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Couper K, Putt O, Field R, Poole K, Bradlow W, Clarke A, et al. Incidence of sudden cardiac death in the young: a systematic review. Br Med J. (2020) 10(10):e040815. doi: 10.1136/bmjopen-2020-040815
2. Priori S, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European society of cardiology (ESC). endorsed by: association for European paediatric and congenital cardiology (AEPC). Eur Heart J. (2015) 36(41):2793–867. doi: 10.1093/eurheartj/ehv316
3. Saarel EV, Law I, Berul CI, Ackerman MJ, Kanter RJ, Sanatani S, et al. Safety of sports for young patients with implantable cardioverter-defibrillators: long-term results of the multinational ICD sports registry. Circ Arrhythm Electrophysiol. (2018) 11(11):e006305. doi: 10.1161/CIRCEP.118.006305
4. Zipes DP, Ackerman MJ, Estes NAM, Grant AO, Myerburg RJ, Hare GV. Task force 7: arrhythmias. J Am Coll Cardiol. (2005) 45(8):1354–63. doi: 10.1016/j.jacc.2005.02.014
5. Christian S, Somerville M, Taylor S, Spence JC, Giuffre M, Atallah J. The impact of physical activity modification on the well-being of a cohort of children with an inherited arrhythmia or cardiomyopathy. Cardiol Young. (2020) 30(5):692–7. doi: 10.1017/S1047951120000803
6. Boisson A, De La Villeon G, Huguet H, Abassi H, Pasquie J, Lavastre K, et al. Physical activity and aerobic fitness in children with inherited cardiac diseases. Arch Cardiovasc Dis. (2021) 114(11):727–36. doi: 10.1016/j.acvd.2021.07.004
7. Reineck E, Rolston B, Bragg-Gresham J, Salberg L, Baty L, Kumar S, et al. Physical activity and other health behaviors in adults with hypertrophic cardiomyopathy. Am J Cardiol. (2013) 111(7):1034–9. doi: 10.1016/j.amjcard.2012.12.018
8. Lang CN, Steinfurt J, Odening KE. Avoiding sports-related sudden cardiac death in children with congenital channelopathy: recommendations for sports activities. Herz. (2017) 42(2):162–70. doi: 10.1007/s00059-017-4549-2
9. Pelliccia A, Fagard R, Bjørnstad H, Anastassakis A, Arbustini E, Assanelli D, et al. Recommendations for competitive sports participation in athletes with cardiovascular disease: a consensus document from the study group of sports cardiology of the working group of cardiac rehabilitation and exercise physiology and the working group of myocardial and pericardial diseases of the European society of cardiology. Eur Heart J. (2005) 26(14):1422–45. doi: 10.1093/eurheartj/ehi325
10. Zipes D, Camm A, Borggrefe M, Buxton A, Chaitman B, Fromer M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death–executive summary: a report of the American college of cardiology/American heart association task force and the European society of cardiology committee for practice guidelines (writing committee to develop guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death) developed in collaboration with the European heart rhythm association and the heart rhythm society. Eur Heart J. (2006) 27(17):2099–140. doi: 10.1093/eurheartj/ehl199
11. Maron B, Thompson P, Ackerman M, Balady G, Berger S, Cohen D, et al. Recommendations and considerations related to preparticipation screening for cardiovascular abnormalities in competitive athletes: 2007 update: a scientific statement from the American heart association council on nutrition, physical activity, and metabolism: endorsed by the American college of cardiology foundation. Circulation. (2007) 115(12):1643–55. doi: 10.1161/CIRCULATIONAHA.107.181423
12. Pelliccia A, Sharma S, Gati S, Back M, Borjesson M, Caselli S, et al. 2020 ESC guidelines on sports cardiology and exercise in patients with cardiovascular disease. Eur Heart J. (2021) 42(1):17–96. doi: 10.1093/eurheartj/ehaa605
13. Cadrin-Tourigny J, Bosman LP, Nozza A, Wang W, Tadros R, Bhonsale A, et al. A new prediction model for ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. (2019) 40(23):1850–8. doi: 10.1093/eurheartj/ehz103
14. Halperin J, Levine G, Al-Khatib S, Birtcher K, Bozkurt B, Brindis R, et al. Further evolution of the ACC/AHA clinical practice guideline recommendation classification system: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. Circulation. (2016) 133(14):1426–8. doi: 10.1161/CIR.0000000000000312
15. Priori S, Wilde A, Horie M, Cho Y, Behr E, Berul C, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. (2013) 10(12):1932–63. doi: 10.1016/j.hrthm.2013.05.014
16. Ackerman M, Zipes D, Kovacs R, Maron B. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: task force 10: the cardiac channelopathies: a scientific statement from the American heart association and American college of cardiology. Circulation. (2015) 132(22):e326–9. doi: 10.1161/CIR.0000000000000246
17. Zeppenfeld K, Tfelt-Hansen J, de Riva M, Winkel B, Behr E, Blom N, et al. 2022 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J. (2022) 43(40):3997–4126. doi: 10.1093/eurheartj/ehac262
18. Schwartz P, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalence of the congenital long-QT syndrome. Circulation. (2009) 120(18):1761–7. doi: 10.1161/CIRCULATIONAHA.109.863209
19. Schnell F, Behar N, Carré F. Long-QT syndrome and competitive sports. Arrhythm Electrophysiol Rev. (2018) 7(3):187–92. doi: 10.15420/aer.2018.39.3
20. Wallace E, Howard L, Liu M, O’Brien T, Ward D, Shen S, et al. Long QT syndrome: genetics and future perspective. Pediatr Cardiol. (2019) 40(7):1419–30. doi: 10.1007/s00246-019-02151-x
21. Tobert K, Bos J, Garmany R, Ackerman M. Return-to-Play for athletes with long QT syndrome or genetic heart diseases predisposing to sudden death. J Am Coll Cardiol. (2021) 78(6):594–604. doi: 10.1016/j.jacc.2021.04.026
22. Schwartz P, Priori S, Spazzolini C, Moss A, Vincent G, Napolitano C, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. (2001) 103(1):89–95. doi: 10.1161/01.CIR.103.1.89
23. Johnson J, Ackerman M. Return to play? Athletes with congenital long QT syndrome. Br J Sports Med. (2013) 47(1):28–33. doi: 10.1136/bjsports-2012-091751
24. Aziz P, Sweeten T, Vogel R, Bonney W, Henderson J, Patel A, et al. Sports participation in genotype positive children with long QT syndrome. JACC Clin Electrophysiol. (2015) 1(1-2):62–70. doi: 10.1016/j.jacep.2015.03.006
25. Chambers K, Beausejour Ladouceur V, Alexander M, Hylind R, Bevilacqua L, Mah D, et al. Cardiac events during competitive, recreational, and daily activities in children and adolescents with long QT syndrome. J Am Heart Assoc. (2017) 6(9):e005445. doi: 10.1161/JAHA.116.005445
26. Chen C, De Souza A, Franciosi S, Harris K, Sanatani S. Physical activity in paediatric long QT syndrome patients - CJC pediatric and congenital heart disease. CJC. (2022) 1(2):80–5. doi: 10.1016/j.cjcpc.2021.12.001
27. Baltogiannis G, Lysitsas D, di Giovanni G, Ciconte G, Sieira J, Conte G, et al. CPVT: arrhythmogenesis, therapeutic management, and future perspectives. A brief review of the literature. Front Cardiovasc Med. (2019) 6:92. doi: 10.3389/fcvm.2019.00092
28. Liu N, Rizzi N, Boveri L, Priori S. Ryanodine receptor and calsequestrin in arrhythmogenesis: what we have learnt from genetic diseases and transgenic mice. J Mol Cell Cardiol. (2009) 46(2):149–59. doi: 10.1016/j.yjmcc.2008.10.012
29. Kazemian P, Gollob M, Pantano A, Oudit G. A novel mutation in the RYR2 gene leading to catecholaminergic polymorphic ventricular tachycardia and paroxysmal atrial fibrillation: dose-dependent arrhythmia-event suppression by β-blocker therapy. Can J Cardiol. (2011) 27(6):870.e7–10. doi: 10.1016/j.cjca.2011.02.003
30. van der Werf C, Kannankeril PJ, Sacher F, Krahn AD, Viskin S, Leenhardt A, et al. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J Am Coll Cardiol. (2011) 57(22):2244–54. doi: 10.1016/j.jacc.2011.01.026
31. Roston T, Vinocur J, Maginot K, Mohammed S, Salerno J, Etheridge S, et al. Catecholaminergic polymorphic ventricular tachycardia in children: analysis of therapeutic strategies and outcomes from an international multicenter registry. Circ Arrhythm Electrophysiol. (2015) 8(3):633–42. doi: 10.1161/CIRCEP.114.002217
32. Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc D, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation. (1995) 91(5):1512–9. doi: 10.1161/01.CIR.91.5.1512
33. Roston T, Yuchi Z, Kannankeril P, Hathaway J, Vinocur J, Etheridge S, et al. The clinical and genetic spectrum of catecholaminergic polymorphic ventricular tachycardia: findings from an international multicentre registry. Europace. (2018) 20(3):541–7. doi: 10.1093/europace/euw389
34. Kurtzwald-Josefson E, Hochhauser E, Katz G, Porat E, Seidman J, Seidman C, et al. Exercise training improves cardiac function and attenuates arrhythmia in CPVT mice. J Appl Physiol (1985). (2012) 113(11):1677–83. doi: 10.1152/japplphysiol.00818.2012
35. Faggioni M, Hwang H, van der Werf C, Nederend I, Kannankeril P, Wilde A, et al. Accelerated sinus rhythm prevents catecholaminergic polymorphic ventricular tachycardia in mice and in patients. Circ Res. (2013) 112(4):689–97. doi: 10.1161/CIRCRESAHA.111.300076
36. Kannankeril P, Shoemaker M, Gayle K, Fountain D, Roden D, Knollmann B. Atropine-induced sinus tachycardia protects against exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. Europace. (2020) 22(4):643–8. doi: 10.1093/europace/euaa029
37. Ostby S, Bos J, Owen H, Wackel P, Cannon B, Ackerman M. Competitive sports participation in patients with catecholaminergic polymorphic ventricular tachycardia: a single center’s early experience. JACC Clin Electrophysiol. (2016) 2(3):253–62. doi: 10.1016/j.jacep.2016.01.020
38. Maron BJ, Udelson JE, Bonow RO, Nishimura RA, Ackerman MJ, Estes NA 3rd, et al., Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: task force 3: hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy and other cardiomyopathies, and myocarditis: a scientific statement from the American heart association and American college of cardiology. Circulation. (2015) 132(22):e273–80. doi: 10.1016/j.jacc.2015.09.032
39. Pelliccia A, Solberg E, Papadakis M, Adami P, Biffi A, Caselli S, et al. Recommendations for participation in competitive and leisure time sport in athletes with cardiomyopathies, myocarditis, and pericarditis: position statement of the sport cardiology section of the European association of preventive cardiology (EAPC). Eur Heart J. (2019) 40(1):19–33. doi: 10.1093/eurheartj/ehy730
40. Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American college of cardiology/American heart association joint committee on clinical practice guidelines. Circulation. (2020) 142(25):e558–631. doi: 10.1161/CIR.0000000000000937
41. James C, Bhonsale A, Tichnell C, Murray B, Russell S, Tandri H, et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. (2013) 62(14):1290–7. doi: 10.1016/j.jacc.2013.06.033
42. Te Riele A, James CA, Sawant AC, Bhonsale A, Groeneweg JA, Mast TP, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy in the pediatric population: clinical characterization and comparison with adult-onset disease. JACC Clin Electrophysiol. (2015) 1(6):551–60. doi: 10.1016/j.jacep.2015.08.004
43. Roudijk RW, Verheul L, Bosman LP, Bourfiss M, Breur J, Slieker MG, et al. Clinical characteristics and follow-up of pediatric-onset arrhythmogenic right ventricular cardiomyopathy. JACC Clin Electrophysiol. (2022) 8(3):306–18. doi: 10.1016/j.jacep.2021.09.001
44. Silka MJ, Shah MJ, Silva JNA, Balaji S, Beach CM, Benjamin MN, et al. 2021 PACES expert consensus statement on the indications and management of cardiovascular implantable electronic devices in pediatric patients: executive summary. Cardiol Young. (2021) 31(11):1717–37. doi: 10.1017/S1047951121003395
45. Cicenia M, Drago F. Arrhythmogenic cardiomyopathy: diagnosis, evolution, risk stratification and pediatric population-where are we? J Cardiovasc Dev Dis. (2022) 9(4):98. doi: 10.3390/jcdd9040098
46. Deshpande SR, Herman HK, Quigley PC, Shinnick JK, Cundiff CA, Caltharp S, et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D): review of 16 pediatric cases and a proposal of modified pediatric criteria. Pediatr Cardiol. (2016) 37(4):646–55. doi: 10.1007/s00246-015-1327-x
47. Landry CH, Fatah M, Connelly KA, Angaran P, Hamilton RM, Dorian P. Evaluating the 12-lead electrocardiogram for diagnosing ARVC in young populations: implications for preparticipation screening of athletes. CJC Open. (2021) 3(4):498–503. doi: 10.1016/j.cjco.2020.12.011
48. Cohen MI, Atkins MB. Arrhythmogenic right ventricular cardiomyopathy in the pediatric population. Curr Opin Cardiol. (2022) 37(1):99–108. doi: 10.1097/HCO.0000000000000937
49. Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: executive summary. Heart Rhythm. (2019) 16(11):e373–407. doi: 10.1016/j.hrthm.2019.09.019
50. Al-Khatib S, Stevenson W, Ackerman M, Bryant W, Callans D, Curtis A, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: executive summary: a report of the American college of cardiology/American heart association task force on clinical practice guidelines and the heart rhythm society. Heart Rhythm. (2018) 15(10):e190–e252. doi: 10.1016/j.hrthm.2017.10.035
51. Gow R, Borghese M, Honeywell C, Colley R. Activity intensity during free-living activities in children and adolescents with inherited arrhythmia syndromes: assessment by combined accelerometer and heart rate monitor. Circ Arrhythm Electrophysiol. (2013) 6(5):939–45. doi: 10.1161/CIRCEP.113.000514
52. Paulin F, Hodgkinson K, MacLaughlan S, Stuckless S, Templeton C, Shah S, et al. Exercise and arrhythmic risk in TMEM43 p.S358L arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm. (2020) 17(7):1159–66. doi: 10.1016/j.hrthm.2020.02.028
53. Ho A, Leach E, Virani A, Arbour L, Bartels K, Wong E. Cascade testing for inherited arrhythmia conditions: experiences and attitudes of family communication approaches for a Canadian cohort. J Genet Couns. (2022) 31(3):815–28. doi: 10.1002/jgc4.1550
54. Lie OH, Dejgaard LA, Saberniak J, Rootwelt C, Stokke MK, Edvardsen T, et al. Harmful effects of exercise intensity and exercise duration in patients with arrhythmogenic cardiomyopathy. JACC Clin Electrophysiol. (2018) 4(6):744–53. doi: 10.1016/j.jacep.2018.01.010
55. Wang W, Tichnell C, Murray BA, Agafonova J, Cadrin-Tourigny J, Chelko S, et al. Exercise restriction is protective for genotype-positive family members of arrhythmogenic right ventricular cardiomyopathy patients. Europace. (2020) 22(8):1270–8. doi: 10.1093/europace/euaa105
56. Wang W, Orgeron G, Tichnell C, Murray B, Crosson J, Monfredi O, et al. Impact of exercise restriction on arrhythmic risk among patients with arrhythmogenic right ventricular cardiomyopathy. J Am Heart Assoc. (2018) 7(12):e008843. doi: 10.1161/JAHA.118.008843
57. Smith E, Lakdawala N, Papoutsidakis N, Aubert G, Mazzanti A, McCanta A, et al. Desmoplakin cardiomyopathy, a fibrotic and inflammatory form of cardiomyopathy distinct from typical dilated or arrhythmogenic right ventricular cardiomyopathy. Circulation. (2020) 141(23):1872–84. doi: 10.1161/CIRCULATIONAHA.119.044934
58. Volpato G, Falanga U, Cipolletta L, Conti MA, Grifoni G, Ciliberti G, et al. Sports activity and arrhythmic risk in cardiomyopathies and channelopathies: a critical review of European guidelines on sports cardiology in patients with cardiovascular diseases. Medicina (Kaunas). (2021) 57(4):308. doi: 10.3390/medicina57040308
59. Shah M. Hypertrophic cardiomyopathy. Cardiol Young. (2017) 27(S1):S25–S30. doi: 10.1017/S1047951116002195
60. Colan S, Lipshultz S, Lowe A, Sleeper L, Messere J, Cox G, et al. Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children: findings from the pediatric cardiomyopathy registry. Circulation. (2007) 115(6):773–81. doi: 10.1161/CIRCULATIONAHA.106.621185
61. Caselli S, Pelliccia A. The electrocardiogram and the phenotypic expression of hypertrophic cardiomyopathy. Eur Heart J. (2019) 40(12):982–5. doi: 10.1093/eurheartj/ehy403
62. Maron M. Clinical utility of cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Cardiovasc Magn Reson. (2012) 14(1):1532–49. doi: 10.1186/1532-429X-14-13
63. Miron A, Lafreniere-Roula M, Steve Fan CP, Armstrong KR, Dragulescu A, Papaz T, et al. A validated model for sudden cardiac death risk prediction in pediatric hypertrophic cardiomyopathy. Circulation. (2020) 142(3):217–29. doi: 10.1161/CIRCULATIONAHA.120.047235
64. O’Mahony C, Jichi F, Pavlou M, Monserrat L, Anastasakis A, Rapezzi C, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J. (2014) 35(30):2010–20. doi: 10.1093/eurheartj/eht439
65. Varnava A, Elliott P, Mahon N, Davies M, McKenna W. Relation between myocyte disarray and outcome in hypertrophic cardiomyopathy. Am J Cardiol. (2001) 88(3):275–9. doi: 10.1016/S0002-9149(01)01640-X
66. Authors/Task Force m, Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task force for the diagnosis and management of hypertrophic cardiomyopathy of the European society of cardiology (ESC). Eur Heart J. (2014) 35(39):2733–79. doi: 10.1093/eurheartj/ehu284
67. Maron B, Olivotto I, Spirito P, Casey S, Bellone P, Gohman T, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation. (2000) 102(8):858–64. doi: 10.1161/01.CIR.102.8.858
68. Konhilas J, Watson P, Maass A, Boucek D, Horn T, Stauffer B, et al. Exercise can prevent and reverse the severity of hypertrophic cardiomyopathy. Circ Res. (2006) 98(4):540–8. doi: 10.1161/01.RES.0000205766.97556.00
69. Corrado D, Basso C, Schiavon M, Thiene G. Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med. (1998) 339(6):364–9. doi: 10.1056/NEJM199808063390602
70. Malhotra A, Sharma S. Outcomes of cardiac screening in adolescent soccer players. N Engl J Med. (2018) 379(21):524–34. doi: 10.1056/NEJMoa1714719
71. Pelliccia A. New evidences recommend an active lifestyle in young HCM patients. Int J Cardiol. (2022) 369:82–3. doi: 10.1016/j.ijcard.2022.08.023
72. Andreassen K, Dejgaard L, Lie Ø, Fink T, Lunde I, Edvardsen T, et al. Exercise training during childhood and adolescence is associated with favorable diastolic function in hypertrophic cardiomyopathy. Int J Cardiol. (2022) 364:65–71. doi: 10.1016/j.ijcard.2022.06.042
73. Sheikh N, Papadakis M, Schnell F, Panoulas V, Malhotra A, Wilson M, et al. Clinical profile of athletes with hypertrophic cardiomyopathy. Circ Cardiovasc Imaging. (2015) 8(7):e003454. doi: 10.1161/CIRCIMAGING.114.003454
74. Chobanian A, Bakris G, Black H, Cushman W, Green L, Izzo J, et al. The seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure: the JNC 7 report. JAMA. (2003) 289(19):2560–71. doi: 10.1001/jama.289.19.2560
75. Luo Q, Chen J, Zhang T, Tang X, Yu B. Retrospective analysis of clinical phenotype and prognosis of hypertrophic cardiomyopathy complicated with hypertension. Sci Rep. (2020) 10(1):1–9. doi: 10.1038/s41598-019-56847-4
76. Mace H, Rizwan A, Lutz W, Hamid A, Campbell W, McMullan M, et al. Hypertension in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. (2023) 81(8):1605. doi: 10.1016/S0735-1097(23)02049-1
77. Drazner M. The progression of hypertensive heart disease. Circulation. (2011) 123(3):327–34. doi: 10.1161/CIRCULATIONAHA.108.845792
78. Pelliccia A, Lemme E, Maestrini V, Di Paolo F, Pisicchio C, Di Gioia G, et al. Does sport participation worsen the clinical course of hypertrophic cardiomyopathy? Clinical outcome of hypertrophic cardiomyopathy in athletes. Circulation. (2018) 137(5):531–3. doi: 10.1161/CIRCULATIONAHA.117.031725
79. O’Mahony C, Tome-Esteban M, Lambiase P, Pantazis A, Dickie S, McKenna W, et al. A validation study of the 2003 American college of cardiology/European society of cardiology and 2011 American college of cardiology foundation/American heart association risk stratification and treatment algorithms for sudden cardiac death in patients with hypertrophic cardiomyopathy. Heart. (2013) 99(8):534–41. doi: 10.1136/heartjnl-2012-303271
80. Pelliccia A, Caselli S, Pelliccia M, Musumeci M, Lemme E, Di Paolo F, et al. Clinical outcomes in adult athletes with hypertrophic cardiomyopathy: a 7-year follow-up study. Br J Sports Med. (2020) 54(16):1008–12. doi: 10.1136/bjsports-2019-100890
81. Olde Nordkamp LR, Postema PG, Knops RE, van Dijk N, Limpens J, Wilde AA, et al. Implantable cardioverter-defibrillator harm in young patients with inherited arrhythmia syndromes: a systematic review and meta-analysis of inappropriate shocks and complications. Heart Rhythm. (2016) 13(2):443–54. doi: 10.1016/j.hrthm.2015.09.010
82. DeMaso DR, Lauretti A, Spieth L, van der Feen JR, Jay KS, Gauvreau K, et al. Psychosocial factors and quality of life in children and adolescents with implantable cardioverter-defibrillators. Am J Cardiol. (2004) 93(5):582–7. doi: 10.1016/j.amjcard.2003.11.022
83. Sears SF Jr, Conti JB. Quality of life and psychological functioning of icd patients. Heart. (2002) 87(5):488–93. doi: 10.1136/heart.87.5.488
84. Lampert R, Olshansky B, Heidbuchel H, Lawless C, Saarel E, Ackerman M, et al. Safety of sports for athletes with implantable cardioverter-defibrillators: results of a prospective, multinational registry. Circulation. (2013) 127(20):2021–30. doi: 10.1161/CIRCULATIONAHA.112.000447
85. Olshansky B, Atteya G, Cannom D, Heidbuchel H, Saarel EV, Anfinsen OG, et al. Competitive athletes with implantable cardioverter-defibrillators-how to program? Data from the implantable cardioverter-defibrillator sports registry. Heart Rhythm. (2019) 16(4):581–7. doi: 10.1016/j.hrthm.2018.10.032
86. Kay B, Lampert R. Devices and athletics: decision-making around return to play. Cardiol Clin. (2023) 41(1):81–92. doi: 10.1016/j.ccl.2022.08.007
87. Lampert R, Olshansky B, Heidbuchel H, Lawless C, Saarel E, Ackerman M, et al. Safety of sports for athletes with implantable cardioverter-defibrillators: long-term results of a prospective multinational registry. Circulation. (2017) 135(23):2310–2. doi: 10.1161/CIRCULATIONAHA.117.027828
88. Ingles J, James C. Psychosocial care and cardiac genetic counseling following sudden cardiac death in the young. Prog Pediatr Cardiol: Prog Pediatr Cardiol. (2017) 45:31–6. doi: 10.1016/j.ppedcard.2017.03.001
89. Cheung C, Laksman Z, Mellor G, Sanatani S, Krahn A. Exercise and inherited arrhythmias. Can J Cardiol. (2016) 32(4):452–8. doi: 10.1016/j.cjca.2016.01.007
Keywords: sports, exercise, physical activity, sudden cardiac death, channelopathy, cardiomyopathy, implantable cardioverter-defibrillator, pediatrics
Citation: Katyal A, Li COY, Franciosi S and Sanatani S (2023) The safety of sports in children with inherited arrhythmia substrates. Front. Pediatr. 11:1151286. doi: 10.3389/fped.2023.1151286
Received: 25 January 2023; Accepted: 22 March 2023;
Published: 4 April 2023.
Edited by:
Patricia Longmuir, University of Ottawa, CanadaReviewed by:
Bruce Alpert, Retired, United StatesEmanuele Micaglio, IRCCS San Donato Polyclinic, Italy
© 2023 Katyal, Li, Franciosi and Sanatani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shubhayan Sanatani c3NhbmF0YW5pQGN3LmJjLmNh
Specialty Section: This article was submitted to Pediatric Cardiology, a section of the journal Frontiers in Pediatrics