 Qianhuining Kuang
Qianhuining Kuang Xu He
Xu He Chunlin Gao
Chunlin Gao Zhengkun Xia
Zhengkun Xia
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 10 July 2023
Sec. Pediatric Nephrology
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1148132
This article is part of the Research Topic Optimizing Outcomes for Children With Immune-Mediated Chronic Kidney Disease View all 10 articles
An eight-year-old girl was admitted with vomiting, gross hematuria, and progressive renal dysfunction. A renal biopsy revealed endocapillary proliferative glomerulopathy and crescent formation. Immunofluorescence staining revealed diffuse granular deposits of IgG and C3. Post-streptococcal acute glomerulonephritis (PSAGN) was suspected, based on the elevated anti-streptolysin O levels, decreased serum C3 concentrations, and histologic findings. The myeloperoxidase anti-neutrophil cytoplasmic antibody (MPO-ANCA) test was positive, and the young patient gradually developed palisaded neutrophilic and granulomatous dermatitis (PNGD), orbital and paranasal sinus granulomatous neoplasms, along with intermittent nose, head, and orbital pain. Finally, she was diagnosed with the rare MPO-ANCA-associated granulomatosis with polyangiitis (GPA) superimposed on PSAGN. The patient was treated with aggressive renal replacement therapy, methylprednisolone pulse therapy, and intravenous pulse cyclophosphamide; her renal function normalized, and her pain symptoms improved.
Antineutrophil cytoplasmic antibody-associated vasculitides (AAVs) are a group of life-threatening primary systemic necrotizing small-vessel vasculitides characterized by the presence of antineutrophil cytoplasmic antibodies (ANCA), including granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA), classified by clinical features. GPA and MPA frequently affect older adults, while EGPA tends to affect young adults. Approximately 60% of AAV cases involve the kidney; this condition is called ANCA-associated glomerulonephritis (AAGN), which can be diagnosed by a renal biopsy and typically manifests as necrotizing and crescentic glomerulonephritis with little to no immunoglobulin (Ig) or complement deposition. Cases progress to end-stage renal disease (ESRD) in 20%–25% of patients, and the renal prognosis of AAV is usually very poor.
Post-streptococcal acute glomerulonephritis (PSAGN) is an infection-related glomerulonephritis (IRGN) caused by group A β-hemolytic streptococci and characterized by endocapillary hypercellularity with immunoglobulin G (IgG) and complement (C3) deposition. Renal damage in PSAGN is caused by the activation of the classical and alternative complement pathways secondary to streptococcal-initiated humoral immunity. In the early stages of PSAGN, serum C3 levels are low and gradually return to the normal range within two months. PSAGN usually has a favorable renal prognosis and requires only suggestive therapy.
In this report, we describe a pediatric case of PSAGN, as suggested by laboratory test results and a renal biopsy, with progressive renal dysfunction that was alleviated by hemodialysis and immunosuppressive treatment. At baseline, MPO-ANCA was detected; during the course of the disease, granulomatous dermatitis and orbital and paranasal sinus granulomas gradually appeared. One case of AAV superimposed on a PSAGN presented with more severe renal dysfunction; however, the exact etiology of this condition remains unknown.
An eight-year-old Chinese girl was admitted with complaints of vomiting, oliguria (150–300 ml/day), and gross hematuria for two days. She had no symptoms of fever, skin infection, or joint pain. Her blood pressure was elevated (122/80 mmHg). Serum laboratory tests revealed mild anemia (hemoglobin 96 g/L), severe acute kidney injury (blood urea nitrogen 35.1 mmol/L, creatinine 564.9 µmol/L, uric acid 610 µmol/L), and abnormal urine tests (185.6 RBCs/HPF, urine protein 0.63 g/24 h). Serum C3 concentration was 0.051 g/L (normal range: 0.9–1.8 g/L), anti-streptolysin O(ASO) was 682 IU/ml, and erythrocyte sedimentation rate (ESR, 54 mm/h) was elevated. Anti-nuclear antibody (1:100), anti-histone antibody+, MPO-ANCA 38.88R U/ml (by ELISA, normal range:<20 RU/ml), p-ANCA (+). Tests for other laboratory indicators, including procalcitonin, C-reactive protein (CRP), tumor markers, anti-cardiolipin antibody, and anti-glomerular basement antibody were negative. CT scans of the chest and abdomen were normal. Renal ultrasonography showed that the size of the right kidney was 115 × 47 mm2, the left kidney was 114 × 46 mm2, and the echo of the renal cortex was enhanced.
A renal biopsy was performed. Light microscopy revealed that among the 22 glomeruli, eight had cellular crescent formation and one had segmental sclerosis (Figures 1A,B). Furthermore, diffuse inflammatory cell infiltration, predominantly neutrophils and macrophages, was observed in the glomerular capillary lumen. The capillary wall appeared to be thickened. The tubulointerstitium displayed moderate acute injury, and the focal brush border of the tubular epithelium was detached. Immunofluorescence staining revealed diffuse granular deposits of IgG and C3 (IgG +, C3 +++) along the mesangial region and glomerular capillary walls (Figures 1C,D). Electron microscopy revealed electron-dense deposits in the mesangium and glomerular basement membrane (GBM) and intermittently in the sub-endothelial region (Figures 1E,F). No sub-epithelial electron-dense deposits were observed. In short, the renal pathology revealed endocapillary proliferative glomerulopathy. Complement component analysis and whole-exon sequence gene detection were normal.
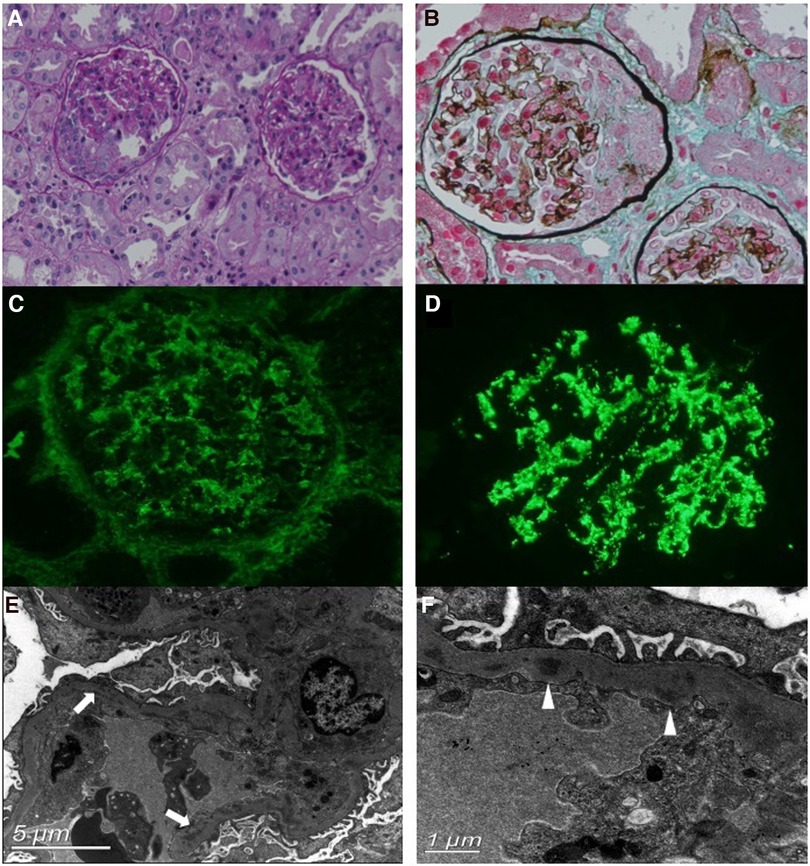
Figure 1. Renal biopsy findings. (A,B) Light microscopy (PAS and Masson's stain, ×200 magnification) shows the cellular crescent formation and diffuse inflammatory cell infiltration. (C,D) Immunofluorescence staining shows diffuse, granular deposits of IgG (C) and C3 (D) along the mesangial region and glomerular capillary walls. (E,F) Electron microscopy reveals electron-dense deposits in the segmental subendothelial region (arrows) and glomerular basement membrane (GBM) (triangles).
Based on the clinical and histologic findings, the patient was diagnosed with PSAGN. However, it seemed to be an atypical case of PSAGN. Given the young patient's rapidly progressive clinical course and extensive crescent formation confirmed by renal biopsy, she was administered two pulses of methylprednisolone (500 mg/day, 20 mg/kg), followed by 40 mg (1.6 mg/kg) of oral prednisone per day, and hemodialysis was performed twice. After 20 days of treatment, serum creatinine levels returned to 29.9 µmol/L, hemoglobin increased to 114 g/L, and urine protein also decreased to 0.1 g/24 h. The serum C3 level was 1.3 g/L. Blood pressure (98/62 mmHg) and increased kidney size (96 × 37 mm2, 100 × 41 mm2) also went back to normal. Considering that the laboratory indicators returned to normal, other immunosuppressive agents were not added temporarily, and oral prednisone was tapered (Figure 2).
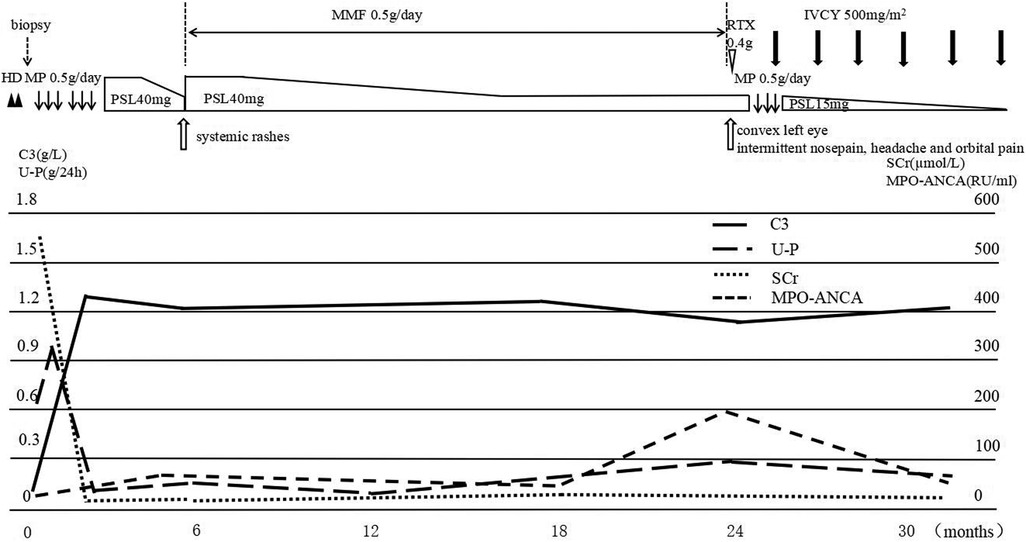
Figure 2. The clinical course of the patient. After hemodialysis and two pulses of methylprednisolone (500 mg/day), followed by 40 mg oral prednisone per day, serum C3 and SCr levels returned to normal, while MPO-ANCA was still positive. Subsequently, the patient developed systemic rashes; prednisone was increased to 40 mg/day, and mycophenolate was prescribed. Two years later, the girl presented with orbital and paranasal sinus abnormalities, the MPO-ANCA level was highly elevated, treatment with one dose of rituximab was unsuccessful, and another pulse of methylprednisolone combined with monthly IVCY brought the disease under control. MP, methylprednisolone pulse; PSL, prednisolone; IVCY, intravenous cyclophosphamide; U-P, urine protein.
Approximately five months later, the patient developed recurrent systemic red rashes (Figures 3A,B). Laboratory tests revealed normal urine tests and renal function, p-ANCA+, MPO-ANCA 71.58 RU/ml, and a skin biopsy showed palisaded neutrophilic and granulomatous dermatitis (PNGD). Therefore, prednisone was added at 40 mg/day, and mycophenolate mofetil (20 mg/kg) was prescribed to control ANCA activation. The rash gradually resolved (Figure 2).
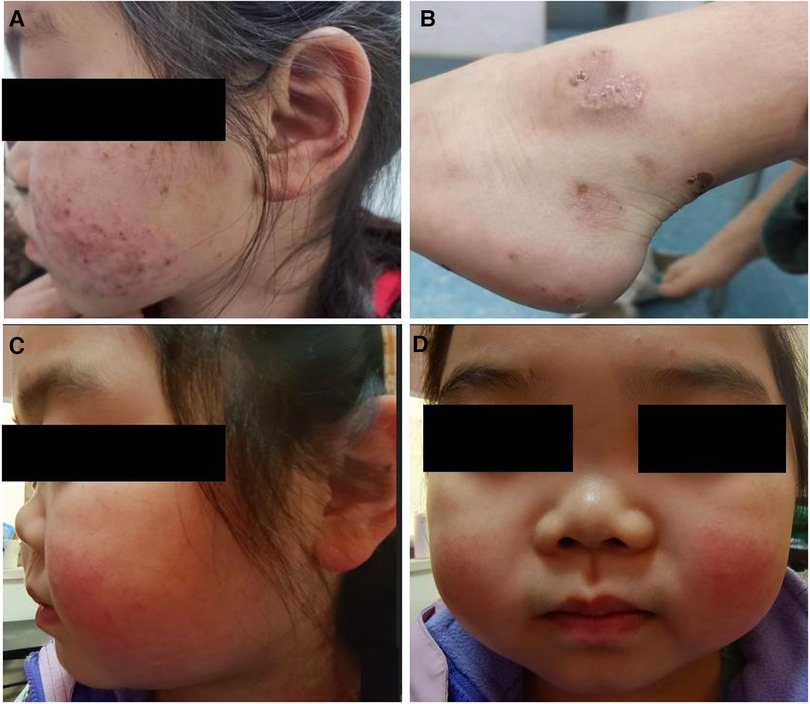
Figure 3. Clinical characteristics of the patient. (A,B) Systemic red rash. (C,D) Cartilage involvement, showing saddle nose deformity.
One and a half years later, the patient presented with a convex left eye and intermittent nose, head, and orbital pain with normal hearing. The bridge of the nose was low and flat, resembling a saddle nose (Figures 3C,D). Imaging revealed orbital pseudotumors and bilateral sinusitis. The patient underwent eye surgery, and the pathology of the left ocular mass indicated chronic inflammation with granulomas and necrosis. We simultaneously prescribed prednisone (15 mg/day), a single dose of rituximab (0.4 g, 375 mg/m2), and suspended mycophenolate mofetil. However, the patient's symptoms of nose, head, and orbital pain did not improve, ESR and CRP were elevated (ESR 38 mm/h, CRP 19.6 mg/L), p-ANCA was still positive, and MPO-ANCA was 193 RU/ml. A sinus biopsy revealed numerous granulomatous neoplasms in both maxillary sinuses. Consequently, the patient was administered one pulse of methylprednisolone (500 mg/day) for three days, followed by oral prednisone at 15 mg/day. Pulsed cyclophosphamide was then administered at 500 mg/m2 per dose for six doses every month. During treatment, the patient's urine and renal function remained normal, and there were no apparent lung lesions. The pain in the nose, eyes, and head gradually subsided.
ANCA is usually thought to be the pathogen in AAV, which is an autoantibody that is directly against the cytoplasm of neutrophils and monocytes and includes mainly cytoplasmic (C-ANCA) and perinuclear (P-ANCA) forms. ANCA is specific to the diagnosis of GPA and MPA. C-ANCA occurs in 95% of new-onset GPA cases, P-ANCA in 80% of new-onset MPA cases, and 40% of new-onset EGPA cases. The prevalence of AAV is approximately 200–400 per million people, and it is rare in children (1). In children, AAV has a higher female preponderance. The peak incidence of AAV occurs in the second decade of life; the median age at diagnosis is 12–14 years; and GPA is more common than MPA or EGPA (2). Clinically, the prognosis of AAV is poor, and immunosuppressive therapy is warranted.
In our case, the clinical course of rapidly progressive abnormal renal function, positive MPO-ANCA at disease onset, and massive crescent formation proven by renal biopsy suggested the possibility of an AAV diagnosis. Subsequently, the girl developed PNGD, orbital and paranasal sinus granulomatous neoplasms, and a saddle nose deformity, accompanied by persistently positive MPO and P-ANCA according to the American College of Rheumatology/European Alliance of Associations (ACR/EULAR) (3) classification criteria, and was diagnosed with GPA. GPA was first described in 1937. The main clinical manifestations of GPA are granulomatous inflammation (orbital pseudotumor, chronic sinusitis, Eustachian tube dysfunction, etc.) and small- or medium-vessel vasculitis (pulmonary hemorrhage, glomerulonephritis, skin purpura, etc.). Upper airway abnormalities, manifesting as chronic rhinitis and serous otitis, may be the earliest presenting features and are present in more than 90% of cases, while abnormalities in the kidneys and lungs are present in 80% and 85% of cases, respectively (4). Historically, GPA has been diagnosed based on the presence of a histopathologic triad including necrotizing angiitis, granulomatous inflammation, and necrotizing crescentic glomerulonephritis (5). The incidence of GPA in the Asian population ranges from 0.37–2.1 per million people per year, and its prevalence in China is approximately 0.194 per million people. Most GPA patients in India and Korea are PR3 positive, while 60% of the GPA patients in China are MPO-ANCA positive (6). In addition, it has been confirmed that the ANCA serotype is more predictive of disease regression and clinical outcomes (7–9). A cohort analysis enrolled 365 patients diagnosed with AAV, 44 (12%) with MPA, and 321 (88%) with GPA. Among the 321 patients with GPA, 273 (85%) had PR3-ANCA, 33 (10%) had MPO-ANCA, and 15 (5%) remained ANCA negative. Compared with MPO-ANCA-positive MPA patients, MPO-ANCA-positive GPA patients were younger at diagnosis, and MPO-ANCA-positive GPA was predominantly female (10). Another case-control study showed that patients with MPO-ANCA-positive GPA were less likely to have severe disease (11) and had lower mortality and higher relapse rates (12). Therefore, it seems that some differences exist between MPO-ANCA-positive and PR3-ANCA-positive patients with GPA in terms of clinical manifestations, they may be different diseases (13).
Finally, the patient was diagnosed with an MPO-ANCA-associated GPA. Although recurrent episodes occurred, no fatal alveolar hemorrhage developed, and renal function quickly returned to normal. These clinical features are consistent with the above observations.
The pathogenesis of AAV is still poorly understood. Two familiar hypotheses are the complementary peptide model and the molecular mimicry model. However, evidence has shown that other immune cell mediators, such as CD4+ T cells, may be involved in AAV onset. Complement activation has been associated with the pathogenesis and progression of AAV. Some studies have reported immunocomplex (IC) formation during the early phases of AAV (14, 15). IC deposition is common in patients with AAGN, and C3 deposition is found in 30%–40% of patients with AAGN and is also an independent risk factor for AAV prognosis (16, 17). The renal biopsy of this patient revealed diffuse granular deposits of IgG and C3 (IgG +, C3 +++), which were incompatible with pauci-immune glomerulonephritis and showed more severe renal involvement in the early stage of the disease course, requiring renal replacement therapy.
ANCA can be caused by environmental exposures (silica), drug use (hydralazine, propylthiouracil, penicillamine, etc.), and disease (chronic inflammatory diseases, neoplasms, and infections). ANCA can also be caused by infection, and the possible mechanisms linking infection and ANCA include the production of neutrophil extracellular traps (NETs) and the ligation of toll-like receptors (TLRs) (18). Infection is associated with the morbidity of various glomerular diseases; the recently proposed concept of streptococcal infection-related nephritis (SIRN) includes IgAN, AAV, PSAGN, and so on. Streptococcal infection and ANCA act synergistically or coincidentally; however, the exact mechanism underlying the streptococcal infection associated with AAV remains unclear. In general, PSAGN patients had a good prognosis in the absence of immunosuppressive therapy. Crescent formation is not rare in PSAGN and are a predictor of poor long-term prognosis (19). In New Zealand children, 41% (11 of 27) of patients with crescentic PSAGN had higher serum creatinine levels, requiring acute dialysis (20). In addition, PSAGN can be associated with many other diseases, such as atypical hemolytic uremic syndrome (aHUS) (21), arthritis (19), IgAN (22), membranous nephropathy (23), and Alport syndrome (22). The co-occurrence of PSAGN and AAV is relatively rare (24, 25). The critical task is to decide whether additional treatment of the ANCA-associated disease is needed or only treatment of the infection. Ardiles et al. (26) tested serum IgG-ANCA levels in 210 patients with PSAGN and 14 patients with streptococcal impetigo without glomerular disease. In the PSAGN group, ANCA was detected in 9% of the patients. However, none of the subjects with streptococcal impetigo tested positive in this study. ANCA was associated with higher serum creatinine levels and more crescent formation, suggesting that ANCA may play a pathogenic role in kidney disease. Therefore, we prescribed glucocorticoids, and the renal function and urine abnormalities recovered quickly. When orbital and paranasal sinus granulomatous neoplasms developed, the combination of prednisone and pulsed cyclophosphamide controlled the disease.
In conclusion, we reported a rare case of MPO-ANCA-associated GPA superimposed on post-streptococcal acute glomerulonephritis, in which the prognosis was relatively good after active hemodialysis and immunosuppressive treatment. In cases of PSAGN with ANCA, although the initial ANCA titer is not very high, we recommend referring to the treatment principle of AAV, and aggressive and long-term maintenance of immunosuppressive therapies is required. A closer follow-up is warranted to focus on extrarenal manifestations and ANCA levels. Given the small number of cases of PSAGN with AAV, the pathophysiologic features and long-term renal prognosis remain unclear.
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding authors.
Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
QK and XH: collecting history, manuscript editing. LJ, ZZ, and CGu: literature search. CGa and ZX: manuscript review. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Almaani S, Fussner LA, Brodsky S, Meara AS, Jayne D. ANCA-associated vasculitis: an update. J Clin Med. (2021) 10(7):1446. doi: 10.3390/jcm10071446
2. Jariwala MP, Laxer RM. Primary vasculitis in childhood: GPA and MPA in childhood. Front Pediatr. (2018) 6:226. doi: 10.3389/fped.2018.00226
3. Robson JC, Grayson PC, Ponte C, Suppiah R, Craven A, Judge A, et al. 2022 American college of rheumatology/European alliance of associations for rheumatology classification criteria for granulomatosis with polyangiitis. Ann Rheum Dis. (2022) 81(3):315–20. doi: 10.1136/annrheumdis-2021-221795
4. Gapud EJ, Seo P, Antiochos B. ANCA-associated vasculitis pathogenesis: a commentary. Curr Rheumatol Rep. (2017) 19(4):15. doi: 10.1007/s11926-017-0641-0
5. Osman MS, Tervaert JWC. Anti-neutrophil cytoplasmic antibodies (ANCA) as disease activity biomarkers in a “personalized medicine approach” in ANCA-associated vasculitis. Curr Rheumatol Rep. (2019) 21(12):76. doi: 10.1007/s11926-019-0872-3
6. Naidu GSRSNK, Misra DP, Rathi M, Sharma A. Is granulomatosis with polyangiitis in Asia different from the west? Int J Rheum Dis. (2019) 22(Suppl 1):90–4. doi: 10.1111/1756-185X.13398
7. Hilhorst M, Arndt F, Joseph Kemna M, Wieczorek S, Donner Y, Wilde B, et al. HLA-DPB1 as a risk factor for relapse in antineutrophil cytoplasmic antibody-associated vasculitis: a cohort study. Arthritis Rheumatol. (2016) 68(7):1721–30. doi: 10.1002/art.39620
8. Cohen Tervaert JW. Should proteinase-3 and myeloperoxidase antineutrophil cytoplasmic antibody vasculitis be treated differently: part 2. Nephrol Dial Transplant. (2019) 34(3):384–7. doi: 10.1093/ndt/gfy406
9. Savige J, Trevisin M, Pollock W. Testing and reporting antineutrophil cytoplasmic antibodies (ANCA) in treated vasculitis and non-vasculitic disease. J Immunol Methods. (2018) 458:1–7. doi: 10.1016/j.jim.2018.02.016
10. Miloslavsky EM, Lu N, Unizony S, Choi HK, Merkel PA, Seo P, et al. Myeloperoxidase-antineutrophil cytoplasmic antibody (ANCA)-positive and ANCA-negative patients with granulomatosis with polyangiitis (wegenercs): distinct patient subsets. Arthritis Rheumatol. (2016) 68(12):2945–52. doi: 10.1002/art.39812
11. Schirmer JH, Wright MN, Herrmann K, Laudien M, Nölle B, Reinhold-Keller E, et al. Myeloperoxidase-antineutrophil cytoplasmic antibody (ANCA)-positive granulomatosis with polyangiitis (wegener’s) is a clinically distinct subset of ANCA-associated vasculitis: a retrospective analysis of 315 patients from a German vasculitis referral center. Arthritis Rheumatol. (2016) 68(12):2953–63. doi: 10.1002/art.39786
12. Mahr A, Katsahian S, Varet H, Guillevin L, Hagen EC, Höglund P, et al. Revisiting the classification of clinical phenotypes of anti-neutrophil cytoplasmic antibody-associated vasculitis: a cluster analysis. Ann Rheum Dis. (2013);72(6):1003–10. doi: 10.1136/annrheumdis-2012-201750
13. Fordham S, Mukhtyar C. Are PR3 positive and MPO positive GPA the same disease? Int J Rheum Dis. (2019) 22(Suppl 1):86–9. doi: 10.1111/1756-185X.13278
14. Hirose O, Itabashi M, Takei T, Honda K, Nitta K. Antineutrophil cytoplasmic antibody-associated glomerulonephritis with immunoglobulin deposition. Clin Exp Nephrol. (2017) 21(4):643–50. doi: 10.1007/s10157-016-1341-1
15. Chen M, Jayne DRW, Zhao MH. Complement in ANCA-associated vasculitis: mechanisms and implications for management. Nat Rev Nephrol. (2017) 13(6):359–67. doi: 10.1038/nrneph.2017.37
16. Oba R, Kanzaki G, Sasaki T, Okabayashi Y, Haruhara K, Okabe M, et al. Long-term renal survival in antineutrophil cytoplasmic antibody-associated glomerulonephritis with complement C3 deposition. Kidney Int Rep. (2021) 6(10):2661–70. doi: 10.1016/j.ekir.2021.08.005
17. Zhang P, Yang X, He X, Hu J, Gao CL, Xia ZK. Relationship between renal damage and Serum complement C3 in children with anti-neutrophil cytoplasmic antibody-associated vasculitis. Nephron. (2021) 145(6):633–41. doi: 10.1159/000516533
18. Konstantinov KN, Emil SN, Barry M, Kellie S, Tzamaloukas AH. Glomerular disease in patients with infectious processes developing antineutrophil cytoplasmic antibodies. ISRN Nephrol. (2013) 2013:324315. doi: 10.5402/2013/324315
19. Ong LT. Management and outcomes of acute post-streptococcal glomerulonephritis in children. World J Nephrol. (2022) 11(5):139–45. doi: 10.5527/wjn.v11.i5.139
20. Wong W, Morris MC, Zwi J. Outcome of severe acute post-streptococcal glomerulonephritis in New Zealand children. Pediatr Nephrol. (2009) 24(5):1021–6. doi: 10.1007/s00467-008-1086-5
21. Parekh M, Konnur A, Gang S. Poststreptococcal glomerulonephritis with atypical hemolytic uremic syndrome: an unusual presentation. Saudi J Kidney Dis Transpl. (2018) 29(3):728–31. doi: 10.4103/1319-2442.235201
22. Suzuki R, Hayashi A, Endo M, Ueda Y, Takahashi T, Sato Y, et al. Cooccurrence of alport syndrome and poststreptococcal acute glomerulonephritis with improvement after steroid administration. CEN Case Rep. (2022) 11(4):453–7. doi: 10.1007/s13730-022-00699-x
23. Matsuda J, Nagayama I, Yamaguchi Y, Itano S, Mori D, Imakita N, et al. A case of crescentic poststreptococcal acute glomerulonephritis (PSAGN) accompanied by membranous nephropathy. Nihon Jinzo Gakkai Shi. (2013) 55(4):567–73.23819387
24. Kanai H, Sawanobori E, Koizumi K, Ohashi R, Higashida K. Pediatric case of crescentic post-streptococcal glomerulonephritis with myeloperoxidase anti-neutrophil cytoplasmic antibody. Clin Nephrol. (2015) 83(4):243–8. doi: 10.5414/CN108286
25. Kamijo N, Mii A, Aratani S, Kashiwagi T, Oda T, Shimizu A, et al. Anti-neutrophil cytoplasmic antibody-associated vasculitis superimposed on post-streptococcal acute glomerulonephritis. Intern Med. (2022) 61(19):2917–23. doi: 10.2169/internalmedicine.8690-21
Keywords: ANCA-associated vasculitides, post-streptococcal acute glomerulonephritis, granulomatosis with polyangiitis, MPO-ANCA, immunosuppressive therapy, immunosuppressive therapy
Citation: Kuang Q, He X, Jia L, Zhang Z, Gui C, Gao C and Xia Z (2023) Case report: A pediatric case of MPO-ANCA-associated granulomatosis with polyangiitis superimposed on post-streptococcal acute glomerulonephritis. Front. Pediatr. 11:1148132. doi: 10.3389/fped.2023.1148132
Received: 19 January 2023; Accepted: 23 June 2023;
Published: 10 July 2023.
Edited by:
Lars Pape, Essen University Hospital, GermanyReviewed by:
Raphael Schild, University Medical Center Hamburg-Eppendorf, Germany© 2023 Kuang, He, Jia, Zhang, Gui, Gao and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunlin Gao c2h1YW5nbXUzNEAxNjMuY29t Zhengkun Xia bmp4emtAMTI2LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.