 Carmela Giancotta1Nicole Colantoni1
Carmela Giancotta1Nicole Colantoni1 Lucia Pacillo1,2
Lucia Pacillo1,2 Veronica Santilli1
Veronica Santilli1 Donato Amodio1Emma Concetta Manno1
Donato Amodio1Emma Concetta Manno1 Nicola Cotugno1,2
Nicola Cotugno1,2 Gioacchino Andrea Rotulo1,3Beatrice Rivalta1,2Andrea Finocchi1,2Caterina Cancrini1,2
Gioacchino Andrea Rotulo1,3Beatrice Rivalta1,2Andrea Finocchi1,2Caterina Cancrini1,2 Andrea Diociaiuti4
Andrea Diociaiuti4 May El Hachem4
May El Hachem4 Paola Zangari1*
Paola Zangari1*
- 1Academic Department of Pediatrics (DPUO), Research Unit of Clinical Immunology and Vaccinology, IRCCS Bambino Gesù Children's Hospital, Rome, Italy
- 2Department of Systems Medicine, University of Tor Vergata, Rome, Italy
- 3Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (DINOGMI), University of Genoa, Genoa, Italy
- 4Dermatology Unit and Genodermatosis Unit, Genetics and Rare Diseases Research Division, IRCCS Bambino Gesù Children's Hospital, Rome, Italy
Inborn errors of immunity associated with atopy (IEIs-A) are a group of inherited monogenic disorders that occur with immune dysregulation and frequent skin involvement. Several pathways are involved in the pathogenesis of these conditions, including immune system defects, alterations of skin barrier and metabolism perturbations. Current technological improvements and the higher accessibility to genetic testing, recently allowed the identification of novel molecular pathways involved in IEIs-A, also informing on potential tailored therapeutic strategies. Compared to other systemic therapy for skin diseases, biologics have the less toxic and the best tolerated profile in the setting of immune dysregulation. Here, we review IEIs-A with skin involvement focusing on the tailored therapeutic approach according to their pathogenetic mechanism.
1. Introduction
Inborn errors of immunity (IEIs) include more than 400 inherited disorders causing specific perturbations of immune development and function (1). The knowledge about the IEIs is increasing over time and it has been definitely demonstrated that severe allergic inflammation may be the initial presentation of the immune system dysregulation. Inborn errors of immunity associated with atopy (IEIs-A) also defined primary atopic disorders (PADs) have been categorized for the first time in 2018 as a subgroup of IEIs characterized by allergy or atopy manifestations (2). Skin involvement is frequently present in these conditions and may present with eczema, urticaria and erythroderma. Among these skin manifestations, atopic dermatitis (AD) is the most common form of eczema, characterized by pruritus, skin inflammation and chronic/relapsing course. IEIs-A diagnosis may be challenging in the setting of skin disorders, and their management and outcome can be widely different. In the last decades, the expanding employment of next-generation sequencing (NGS) has resulted in the identification of novel candidate disease genes and enabled the molecular diagnosis of an increasing number of patients with IEIs. The identification of specific gene defects in IEIs-A has the opportunity to inform on possible therapeutic targets and personalized approaches.
In the present review, we focus on IEIs-A with skin manifestations and in particular on their pathogenetic mechanism and the therapeutic approach targeting the underpinning immune defect. Literature review was performed using Pubmed, Scopus,Web of Science databases and ClinicalTrials.gov, recovering publications on IEIs with atopic manifestations. The search approach was performed using a free-text search (keywords: inborn errors of immunity, primary immunodeficiency with atopy and allergy, atopic disorders, tailored therapies, biologic drugs). We searched recent articles published up to December 2022.
2. Pathogenetic mechanism and treatment of IEIs-A with skin involvement
IEIs-A include different genetic disorders with several pathogenetic pathways responsible for generating an atopic environment, possibly associated with elevation of serum total immunoglobulin (Ig) E. The major mechanisms involved in the genesis of atopy, range from immune system defect and alterations of skin barrier to metabolism perturbations (Table 1).
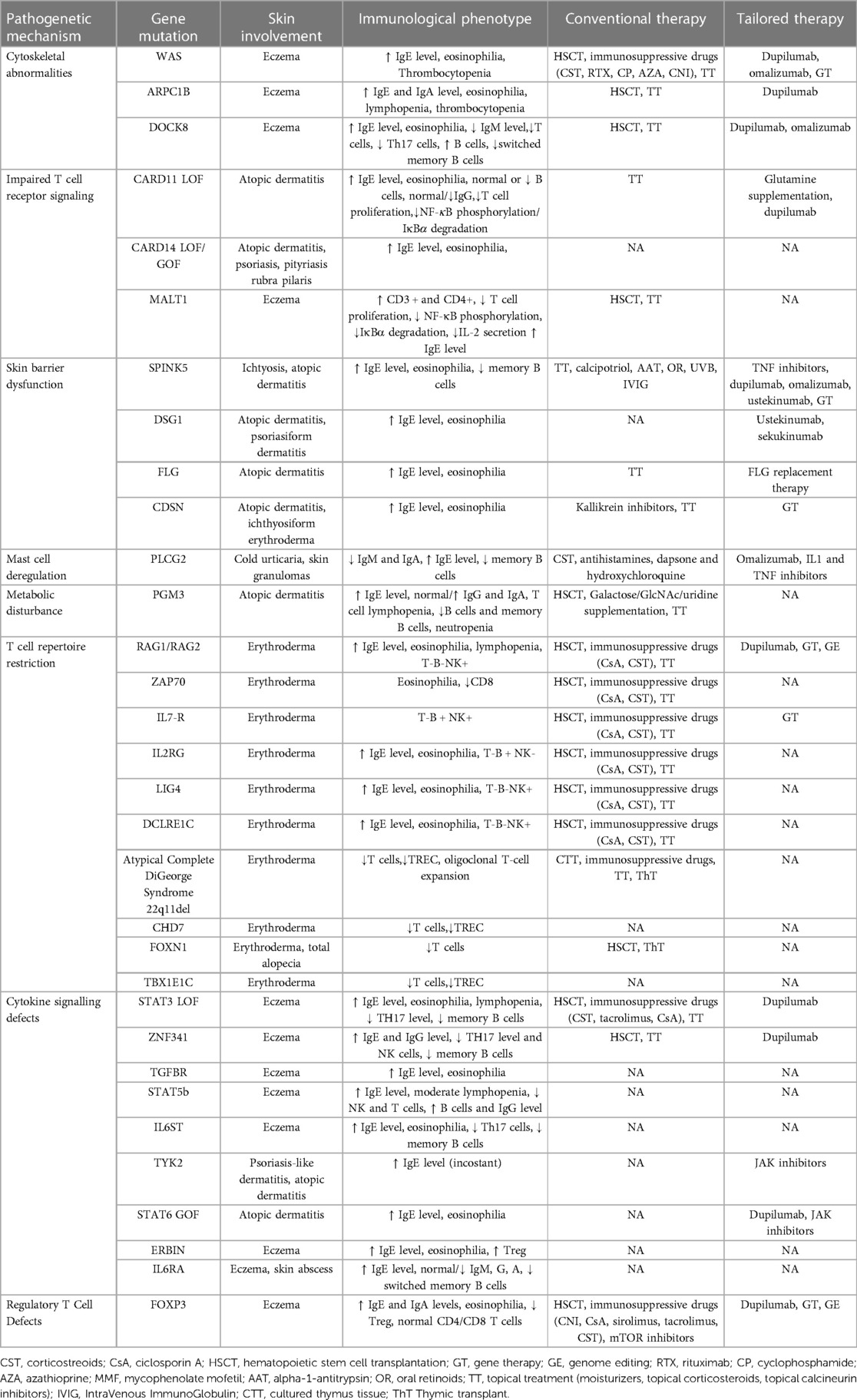
Table 1. Classification of IEIs-A.
2.1. Impaired T cell receptor signaling and cytoskeletal remodeling
2.1.1. WAS
Wiskott-Aldrich syndrome (WAS) is an X-linked recessive disorder characterized by thrombocytopenia, infections, eczematous rash and a high risk of developing malignancy and autoimmune diseases. The illness is due to mutations in WAS gene which encodes WAS protein (WASp), involved in cell signaling and remodeling of cytoskeleton in hematopoietic cells. The WASp is crucial for T cell proliferation, differentiation and survival. WASp deficient T lymphocytes show gene transcription alterations of Th1 cytokines, leading to a skewed Th2 response (3). The restriction of T cell receptor-repertoire diversity has been shown to contribute to this immune dysregulation (4). WAS patients present normal frequency of regulatory T (Treg) cells, but their function is impaired as demonstrated by low interleukin (IL)-10 production potentially predisposing to pathological inflammation and autoimmunity (5). It has been reported that WASp is also involved in the development of regulatory B (Breg) cell, affecting the equilibrium and migration of Treg and Th17 cells during the inflammatory state (6). High levels of Th2 and Th17 cytokines have been found in the skin, as well as the itch-associated molecules (7) both contributing to an inflammatory environment. Moreover, high eosinophils and serum IgE levels are often present (3).
Eczema is found in about 80% of WAS patients, with the characteristics of an early onset, severe and widespread AD, accompanied by petechiae and purpura due to the associated thrombocytopenia.
WAS treatment strategies consist of supportive measures, hematopoietic stem cell transplantation (HSCT) and gene therapy. Immunosuppressive/immunomodulatory drugs to control autoimmune diseases linked to WAS include corticosteroids, intravenous immunoglobulin (IVIG), rituximab, cyclophosphamide, azathioprine, and calcineurin inhibitors (8). With regard to the dermatitis the treatment is based on topical emollients, corticosteroids and according to some authors antiseptic baths (3).
Immunosuppressive drugs (corticosteroids, cyclosporine) are usually administered to control immune dysregulation signs. Anakinra, a human IL-1 receptor antagonist, has been administered with good results, suggesting an involvement of the innate immunity in the generation of auto inflammatory manifestations in WAS patients (9). It has been also described a partial response to omalizumab, a humanized recombinant monoclonal anti IgE antibody, in a genetically confirmed child with WAS and atypical clinical manifestations. The patient presented a history of diffuse pruritic eczema resistant to conventional systemic immunosuppressive therapy, which improved after three subcutaneous injections of omalizumab with concomitant topical steroid (10).
Patients with classic WAS are prone to autoimmune disorders and lymphoma or other malignancies (11). However, the clinical phenotype of WAS is variable and there are patients with less severe symptoms who survive childhood and therefore do not require transplantation, especially in cases due to hypomorphic variants in the WAS gene (12). In the classic form of WAS, the gold-standard treatment is represented by bone marrow transplantation (13–15). The outcomes of children undergoing HSCT are optimal, with a survival rate of more than 97%. In contrast, the few patients who did not underwent HSCT did not reach adulthood (16). Age at HSCT seems to be the only factor significantly associated with overall survival (OS), in fact, patients below 5 years of age have higher OS compared with those who were older then 5 years at the time of HSCT (5-year OS: 94% vs. 66%, respectively) (14). Conversely, OS is not significantly associated with conditioning regimen intensity, donor type, hematopoietic cell source, disease severity, and WASp expression. Full chimerism seems to decrease the incidence of post-HSCT autoimmune diseases and chronic inflammation. Of note, some clinical features of the syndrome, such as AD, may persist in a percentage of patients after transplantation (17).
It was recently described a WAS patient who developed graft-versus-host disease (GvHD) following HSCT. Skin lesions and a high titer of IgE persisted after the use of immunosuppressive treatment. Therefore, a Th2 pathogenesis has been hypothesized, and dupilumab, a monoclonal antibody that inhibits IL-4 and IL-13 signaling, was started with significant clinical benefit (18).
Gene therapy is another effective and safe treatment for WAS providing an adequate immunological reconstitution and control of autoimmunity in most patients (13). Currently, the use of lentiviral vector gene therapy showed great efficacy in patients with WAS who do not have a compatible donor (19).
2.1.2. ARPC1B
Atopic manifestations are described in the platelet abnormalities with eosinophilia and immunomediated inflammatory disease (PLTEID) due to biallelic variants of the actin-related protein 2/3 complex subunit 1B (ARPC1B) gene. PLTEID patients present a broad spectrum phenotype resembling WAS phenotype including severe inflammatory state, lymphoproliferation, purpura, bleeding and immunodeficiency characterized by eczema, severe infections and early-onset vasculitis (20).
ARPC1B protein is a component of the actin-related protein 2/3 complex (Arp2/3) and together with WASp regulate cytoskeletal remodeling of actin and the DNA damage response (21).
Auto inflammatory manifestations of ARPC1B patients are potentially controlled by immunosuppressive therapy such as corticosteroids, mofetil mycophenolate, and rapamycin. Conversely, the use of anti-TNF drugs led to unsatisfactory results. Given the early onset symptoms and the severity of comorbidities, HSCT is currently the only curative treatment (22). In seven ARPCB1 patients, allo-HSCT has been associated with a high survival rate with limited post-transplant morbidity (23). At a median follow-up of 19 months, 6 out of 7 patients are alive and disease-free.
In selected cases, specifically in presence of atopic disorders, biological drugs targeting Th2 pathway could be used. We recently reported a substantial improvement of eczema after starting dupilumab in an ARPC1B child whose phenotype was characterized by frequent infections, thrombocytopenia, elevated eosinophils, IgA and IgE levels, vasculitis, colitis and severe dermatitis refractory to conventional medical therapy. At the age of 10 years, she received dupilumab with significant improvement of dermatitis and itchiness (21).
2.1.3. DOCK8
Dedicator of cytokinesis 8 (DOCK8) encodes a protein highly expressed in lymphocytes behaving as actin cytoskeleton regulator. Biallelic loss-of-function (LOF) DOCK8 mutations result in a combined immunodeficiency characterized by atopy, severe infections, autoimmunity, and malignancy. DOCK8 deficiency impairs the survival, function and migration of immune cells and it impacts both innate and adaptive immune responses. Adaptive immune response is affected through several mechanisms. Among them the main mechanism is related to the impaired actin cytoskeleton rearrangement that causes a defective immune synapse formation. This contributes to impaired B, T and NKT cell survival and long-lived memory responses. Moreover, NK and CD8 cells show an impaired effector activity (24). Naïve DOCK8-deficient CD4+ T cells display increased differentiation towards the Th2 cells and a higher proportion of activated cells producing Th2 cytokines when compared to controls (24).
Few cases of pediatric patients with DOCK8 mutation treated with dupilumab are described so far. Two female patients, 10 and 11.5 years old respectively, obtained a substantial clinical benefit from dupilumab administration after only one month of treatment. The itchiness was much improved and also secondary skin infections were reduced, without increase in systemic infections. Serum IgE levels decreased significantly after treatment (25).
The use of omalizumab in an adult patient with DOCK8 mutation has been described with an improvement of skin lesions (26).
Biological drugs are a viable alternative to improve the skin manifestations, and consequently the quality of life, in patients awaiting HSCT, that remains the only resolutive treatment.
An international survey of 136 DOCK8 transplanted patients patients showed an OS of 87% at 10 years, 47% at 20 years, and 33% at 30 years (27). A multicenter retrospective study of 22 patients reported an OS of 89% after matched related HSCT and 81% after unrelated HSCT (28).
2.1.4. MALT1
Mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) is a paracaspase assembled with B cell CLL/lymphoma 10 (BCL10). Following receptor stimulation, BCL10-MALT1 binds to a caspase recruitment domain (CARD) family proteins such as CARD9, CARD10 or CARD11, forming the CARD-BCL10-MALT1 (CBM) complex. It binds antigen receptors activating the signaling of the NF-κB, JNK, and mTORC1 pathways. The CBM complex and consequently MALT1, have a crucial role in activation, survival, proliferation and metabolism of lymphocytes. Germline LOF variants in MALT1 clinically present recurrent infections, oral and intestinal mucosal involvement, dermatitis and failure to thrive. The impaired CARD-dependent signaling observed in keratinocytes of MALT1 deficient patients could alter the skin barrier and lead to an increase risk of skin infections as well as dermatitis (29). Since the relevance of the CBM complex in the development of several diseases, targeted drugs acting on this pathway are recently attracting research interest. In particular, MALT1 inhibitors are considered specific and efficient drugs that might be finally promising options for the therapy of malignancies and diseases associated with lymphoproliferation (30).
Of note, MALT1 deficiency has been successfully treated with HSCT (31–34).
2.1.5. CARD11
CARD11 is a multidomain scaffold protein needed to induce NF-kB, JNK, and mTOR following antigen receptor stimulation. Germline CARD11 mutations are mainly associated to three different IEIs: CARD11 deficiency, B cell expansion with NF-kB and T cell anergy (BENTA) and CARD11-associated Atopy with Dominant Interference of NF-kB Signalling (CADINS). CADINS is due to heterozygous LOF dominant negative variants of the gene. Many mutations associated with CADINS also downregulate TCR-mediated mTORC1 activation, probably due to a reduction in the glutamine uptake. TCR signaling abnormalities cause an impaired T cell proliferation/activation, an increase of Th2 cytokines and a decreased of Th1 cytokines production. Clinically, patients with CADINS usually present early onset atopy (AD, asthma, food allergies, and eosinophilic esophagitis), recurrent viral skin and respiratory tract infections (35). In a recent single center cohort study, AD and skin infections ameliorated or even resolved during adolescence, suggesting a spontaneous dermatological improvement over time (36).
Glutamine is involved in immunomodulatory functions, but its impact on regulating T-cell function is still unclear (37). In infants with low birth weight and atopy, glutamine supplementation has been studied with a promising reduction in AD (38). CARD11 mutations prevent the upregulation of the glutamine transporter ASCT2 and mTORC1 activating cell proliferation, thus glutamine supplementation has been proposed to improve atopic manifestations in CARD11 or related genes mutations. Interestingly, amino acid supplementation could modulate the immune metabolism and also improve AD (39).The Th2/Th1 imbalance in CARD11 deficient patients indicates that dupilumab might be useful in controlling AD. Case reports of CADINS patients with severe AD successfully treated with dupilumab, without side effects, have been described (40–42).
2.1.6. CARD14
CARD14 induces the NF-κB and mitogen-activated protein kinase (MAPK or MAP kinase) signaling through BCL10 and MALT1, upregulating pro-inflammatory genes. While upregulation of CARD14 gives a skin picture overlapping with psoriasis (43) and atypical juvenile pityriasis rubra pilaris (44, 45), its downregulation is associated with AD, increased risk of skin infection and also dysregulating cutaneous inflammation. Dominant LOF mutations in CARD14 are associated with severe AD, impaired NF-kB cascade, and dysregulation of innate immunity mediators involved in AD pathogenesis (46). The upregulation of CARD14 lead to excessive expression of NF-kB-responsive genes and initiate the recruitment of the inflammatory cells, including dendritic cells and T cells producing IL-23 and IL-17/IL-22 respectively (47). In line with this, ustekinumab, an inhibitor that targets both IL-12 and IL-23 cytokines, proved to be a successful treatment in an increasing numbers of patients with CARD14 GOF mutations (45, 48–51). Since the role of CARD14 in both AD and psoriatic diseases, targeted therapies in these patients need to be considered with caution. To our knowledge, there are no clinical reports on the application of targeted therapy in patients with CARD14 LOF mutations.
2.2. Skin barrier dysfunction
2.2.1. SPINK5
The Comèl-Netherton syndrome (NS) is an inherited disease due to biallelic mutations in the serine protease inhibitor Kazal-type 5 (SPINK5) gene, encoding for inhibitor lympho-epithelial Kazal-type-related inhibitor (LEKTI) that regulates many proteolytic events including the cleavage of desmosomal connections (52). SPINK5 is necessary to maintain skin barrier integrity. In fact, LEKTI deficiency results in defective barrier function (1). NS patients may present with erythroderma or ichthyosis linearis circumflexa as well as typical hair anomalies called trichorhexis nodosa (bamboo hair) (53). Skin and hair defects persist over time, but the disorder usually ameliorates with age (52). The mortality rate is high in the first years of life due to potentially fatal complications (54). In NS epidermidis, the exaggerated protease activity causes the overexpression of proinflammatory, and proallergic cytokines. These molecules drive Th2 cytokine production and lead to atopy and elevated IgE level (55, 56). The IL-17/IL-23 pathway was found to be a predominant signaling axis in NS (57). Recently, 2 endotypes of NS are distinguished on the basis of multiomics analysis: NS with typical ichthyosis linearis circumflexa (NS-ILC) and scaly erythroderma (NS-SE). In NS-ILC, a Th2- complement driven immune response was observed with neutrophil infiltration and complement activation. In NS-SE, a type I IFNγ-driven inflammatory axis appeared prevalent (58).
Conventional treatments include skin care along with supportive care. These interventions can improve cutaneous symptoms without restoring the skin integrity entirely. The use of low-potency corticosteroids, as well as topical calcineurin inhibitors, have shown beneficial effects in some patients (55) but, due to the barrier defect, significant cutaneous absorption cannot be ensured. Over the years, several groups described a significant improvement of the cutaneous signs and symptoms in NS children treated with monthly IVIG (59–64). It was hypothesized that IVIG treatment in NS patients decrease the inflammation by downregulating type 17 inflammation and restoring immune homeostasis (65). Therefore, a trial of IVIG may be considered in severe NS patients.
The administration of kallikrein inhibitors consists of a protein replacement therapy and would be a more etiological treatment. It appears to ameliorate symptoms of NS in animal models (66) and promising results have been observed also in humans (67).
Gene therapy using a lentiviral vector encoding the SPINK5 gene is under investigation (68, 69). A recent trial proposed grafting autologous epidermal sheets derived from genetically modified skin stem cells to release LEKTI protein in NS patients. Results are still not available (70).
Several case reports showed a clinical improvement in adults and children treated with dupilumab (66, 71–73). Currently, a randomized clinical trial evaluating the efficacy and safety of dupilumab in NS patients is under recruitment (74).
Omalizumab administration reduced skin and mucosal symptoms in a 20-year-old patient with NS (75).
A significant skin improvement was shown in a young adult with NS who received ustekinumab (57). Furthermore, promising results came from the use of monoclonal antibodies against IL- 17 called ixekizumab and secukinumab, TNF-α inhibitors (eg, infliximab) and anakinra to control the inflammatory skin lesions in NS (65, 76–78). The different therapeutic responses observed after inhibition of the IL-17 axis suggest that different pathways may contribute to NS pathogenesis (79). Finally, the recent discovery of two NS endotypes could inform new therapeutic approaches (58).
3. Other genes
Filaggrin, a filament-aggregating protein (FLG) is a key protein of the stratum corneum. LOF FLG mutations or mutations causing a decrease in FLG copy number are strongly associated with AD confirming its fundamental function for epidermal barrier integrity (80). Recently, LOF variants in the FLG gene has been recognized as a risk factor for the onset of severe manifestations of food allergy (81). FLG replacement treatment studies in murine models evidenced beneficial effects (82–84).
Desmoglein 1 (DSG1) protein belongs to the family of cadherins. Biallelic LOF mutations in DSG1 gene cause severe dermatitis, multiple allergies and metabolic wasting (SAM) and can manifest as ichthyosiform erythroderma at birth (85). Based on an IL-17–skewed inflammatory signature revealed in these patients, the use of anti IL-17 A antibody and an IL-12/IL-23 antagonist have been proposed with promising results (86, 87).
Corneodesmosin (CDSN) is necessary for cell adhesion and skin integrity. Its expression is reduced in AD patients (88). Peeling skin syndrome (PSS) type B is a rare autosomal recessive disease caused by mutations in the CDSN gene. It is characterized by congenital ichthyosiform erythroderma and skin exfoliation along with elevated serum IgE. The use of antihistamines and kallikrein inhibitors have been proposed based upon the observation in vitro that histamine attenuates the expression of desmosomal proteins in human keratinocytes, and kallikreins are upregulated in type B PSS (89). Recently, in vitro studies for a protein replacement therapy in PSS patients showed encouraging results (90).
3.1. Mast cell deregulation
Germline mutations in two different genes called PLCG2 and ADGRE2 which encode for Phospholipase C gamma 2 and Adhesion protein-coupled receptor E2 respectively, are associated with a type of urticaria triggered by cold and vibration. Mutations in the PLCG2 gene are associated to PLCG2-associated antibody deficiency and immune dysregulation (PLAID) and to auto inflammation and PLCG2-associated antibody deficiency with immune dysregulation (APLAID) syndrome. PLAID syndrome is characterized by early-onset cold urticaria, antibody deficiency, recurrent infections, autoimmune disease and symptomatic allergic disease (91–93). Patients with PLAID, when possible, should avoid cold triggers. Systemic corticosteroids seem to improve symptoms and partially control the disease. In addition, the use of other drugs such as antihistamines, omalizumab, dapsone, and hydroxychloroquine shows improvement in skin symptoms (94). As a future option, the use of specific inhibitors to normalize PLCG2 function at body temperature and to avoid uncontrolled activation at cold exposure has been proposed, but no data are available (92).
3.2. Metabolic disturbance
Hypomorphic phosphoglucomutase 3 (PGM3) mutations with autosomal recessive transmission cause abnormal protein glycosylation and differences in the cellular metabolism. The clinical presentation is characterized by high serum IgE, atopy, neurological impairment, immunodeficiency and autoimmunity (95). In fact, it was demonstrated that altered glycosylation due to PGM deficiency may also affect a subset of lymphocytes (96, 97).
Substrate supplementation therapies for the defective glycosylation pathway have been proposed for several congenital disorders (98). As future therapy perspectives, a trial administering N-acetylglucosamine and uridine oral supplementation to patients with PGM3 deficiency is still on going (99).
HSCT is known to be a curative treatment for most immunodeficiencies, but data in these conditions are limited. Two out of three children described by Pedersen et al. were successfully transplanted, while the other patient died due to infectious complications before transplantation (97).
3.3. T cell repertoire restriction
Omenn syndrome (Os) is an atypical presentation of severe combined immunodeficiency (SCID) with early-onset severe erythroderma and eczema, alopecia, lymphadenopathy, hepatosplenomegaly, chronic persistent diarrhea, recurrent infection and growth failure.
Many genetic defects responsible for TCR over activation can cause uncontrolled lymphocyte expansion and subsequently lymphocyte peripheral infiltration in various tissues including skin, causing tissue damage. Expansion of T-cell clones in Os is associated to Th2 differentiation, Th2 cytokines production, high levels of IgE and eosinophilia. Histological findings in erythroderma of Os are analogous to those described in severe AD. The mechanisms underlying the immunological alterations responsible for the atopic features observed in Os are still a matter of debate. Lymphopenia-induced homeostatic proliferation, poor thymic control of autoreactive lymphocytes, defective Treg and Th2 skewed response have been reported (100).
Since Os is fatal in early life, HSCT represents the first line therapy (101).
In these patients, immunosuppressive treatments such as cyclosporine and steroids are administered as bridging therapy pending HSCT. Cyclosporin, compared to steroids, can modulate T-cell functions at low concentrations, with a consequent control of immune reactivity and skin improvement (102). Finally, immunosuppression provides control of self-reactive T cells but it is often associated to side effects (103, 104). Targeted therapies downregulating Th2 response are considered as new and safe candidates for Os management. Recently, an in vitro model with Os CD4+ T cells showed only a mild reduction of IL-4 production after dupilumab treatment vs. control (105). This could suggest that Th2 polarized response in Os patients might not be regulated by IL-4 signaling only.
Autologous stem-cell-based gene therapy represents the new therapeutic option to treat Os patients without suitable donors. Murine models with RAG mutations treated with lentivirus-mediated gene therapy showed both immunological and clinical improvement, with a dramatic increase in naïve T cells and reduction in effector/memory T cells, and a decrease in cellular infiltration in the skin (106, 107). Currently, preclinical studies are on going to implement transgene expression and obtain stable immune reconstitution (108, 109).
3.4. Cytokine signaling defects
3.4.1. STAT3
The prototypic hyper-immunoglobulin E syndrome (HIES) is caused by LOF autosomal dominant mutations in the signal transducer and activator of transcription 3 (STAT3).
STAT3 activity is essential in several immunological functions including differentiation of Th17 lymphocytes. STAT3 is a transcription factor modulating expression of various genes including cytokines involved in multiple pathways such as IL-6, IL-21, IL-10, IL-11, IL-22, and IL-23. This aberrant immunological transduction explains the various manifestations involving multiple organs and systems, including eczema, lung disease, skeletal and connective tissue abnormalities and vasculopathy. Indeed, the infectious phenotype in patients with STAT3 deficiency is characterized by recurrent staphylococcal skin infections, recurrent bacterial pneumonia and chronic mucocutaneous candidiasis. Interestingly, despite high IgE levels, patients have low rates of allergy and anaphylaxis due to lower affinity of IgE for allergens (110). The skin involvement differs from common AD for early onset and other characteristic signs, such as hyperkeratosis of facial skin, retro auricular fissures, and severe folliculitis (111).
STAT3-deficient patients benefit antibiotic prophylaxis to prevent both dermatological and pulmonary infection. Antifungal prophylaxis should be considered in patients with structural airway abnormalities (110). IVIG replacement showed a decrease in frequency of bacterial pneumonia and can be considered to prevent recurrent lung infection (112).
Published data on HSCT in STAT3 patients are limited and controversial. In the past years results were not encouraging, with reports of transplant failure and death (113–115).
However, recent case series and follow-up studies demonstrated clinical improvement in terms of skin and pulmonary symptoms and immunological reconstitution after HSCT (116, 117).
Conventional therapy for skin manifestations includes topical and systemic immunosuppressive drugs such as steroid, tacrolimus, and cyclosporine. Given the increased IL-4 expression observed in patients with dominant negative STAT3 mutations, it was supposed that dupilumab might treat some clinical manifestations (118). Many reports confirmed the success of the treatment with substantial improvement of the cutaneous lesions, pruritus and IgE levels (118–121).
Omalizumab demonstrated its efficacy in many immune-mediated and autoimmune skin disorders, although its role in HIES is still being defined. Several case reports described its use in STAT3 deficient patients with successfully improvement of skin symptoms and a decrease of serum IgE during treatment (26, 122, 123). Some clinical experience also reported an improvement of pulmonary manifestations (124, 125). Omalizumab was also used in combination with co-trimoxazole and inhaled tobramycin with no recurrent pulmonary or skin infection and a considerable improvement in skin lesions (126).
3.4.2. ZNF341
Patients with Zinc Finger Protein 431 (ZNF341) deficiency phenotypically overlap with STAT3 deficiency. However, patients with ZNF341 deficiency are characterized by less severe non-hematopoietic phenotypes and more severe inflammatory manifestations compared to STAT3 deficiency (118). Patients with increased radiosensitivity and subsequent increased risk of malignancy are reported (127). Furthermore, ZNF341 deficiency seems to influence several immune cells including monocytes and NK lymphocytes, which could contribute in the generation of atopic eczema.
It was reported significantly clinical improvement and reduced IgE level in a ZNF341 deficient adult patient with severe AD following dupilumab administration (128)..
3.4.3. TYK2
Tyrosine kinase 2 (TYK2) enzyme is a member of JAK family and is implicated in the signal transduction of many cytokines including IFN-α, IL-10, IL-6 and IL-12. TYK2 deficiency was discovered in patients with autosomal recessive (AR) HIES. Interestingly, unlike the first patient reported, the HIES phenotype was not found by the other seven patients with TYK2 deficiency described so far (129). However, it seems that various mutation types may influence the expression of TYK2 and promote Th2 cell differentiation, resulting in increased production of Th2 cytokines (130).
4. Novel genes associated with cytokine signaling defects
Since the advent of NGS, a growing number of mutations associated with cytokine signaling defects are being identified but data on targeted therapy are not yet available (131–133).
STAT5 is essential for mast cell cytokines production, proliferation and survival. The role of STAT5B in the IgE-mediated mast cell function has been confirmed in murine models (134).
STAT6 mediates the pathway of IL-4 and a hyperactive STAT6 signaling may alters many cellular processes including increased Th2 differentiation, Th2 cytokines production, elevated IgE levels, increased expression of receptor CD23 on B cells, recruitment of eosinophils and mast cells. This immune dysregulation causes allergic inflammation, asthma and AD (135). Two recent papers identified heterozygous GOF variants in STAT6 characterized by early onset al.lergic phenotype, refractory AD, hyper eosinophilia, high levels of IgE and vascular anomalies of the brain (136, 137). Since the involvement of the IL-4 axis was demonstrated, the use of dupilumab could be a valid therapeutic option. One of the patients reported is currently treated with dupilumab with good clinical outcome. Moreover, the authors demonstrated that in vitro JAK inhibition through ruxolitinib and tofacitinib effectively contained the increased STAT6 phosphorylation in cells expressing the STAT6 variants, proposing JAK inhibitors as a valid therapeutic approach in patients with GOF STAT6 variants (136).
Variants in genes encoding the transforming growth factor β (TGFβ) receptor, cause Loeys–Dietz syndrome (LDS), a rare connective tissue disorder that affects the heart, blood vessels, eyes, and skeletal system. Recently, an allergic phenotype characterized by asthma, food allergy, allergic rhinitis and atopic eczema, has been described (138). LDS mutations appear to lead lymphocytes to acquire and/or maintain Th2 effector functions. It was demonstrated that patients with mutations in TGFβ showed raised levels of IgE and mild reduced IL-17 cytokine production (139).
ERBB2-interacting protein (ERBIN) is necessary for TGF-β pathway activation and its expression is related to STAT3 signaling. In fact, reduced ERBIN expression was described in patients with STAT3 mutations. A LOF ERBIN mutation was recently reported causing Treg and Th2 polarization and a pathological phenotype overlapping with LDS and STAT3-HIES (140).
Many variants of the Interleukin 6 Signal Transducer (IL6ST) gene associated with a severe AR HIES have been identified (141). Indeed, IL6ST gene encodes for a co-receptor of IL-6 called GP130, which transduces the STAT3 pathway (142). Despite clinical phenotype similarities, unlike STAT3 deficiency, AR IL-6R deficiency does not show skeletal involvement (143).
7. Regulatory T Cell DefectsIPEX syndrome is an X-linked autoimmune disease caused by mutations in forkhead box P3 (FOXP3) gene. The clinical phenotype mainly includes immune dysregulation, polyendocrinopathy and enteropathy (144). FOXP3 protein is implicated in the regulation and function of Treg cells, which mediates the suppression of autoreactive T cells (145). Impaired FOXP3 expression leads to a Th2-skewed predominance. Skin involvement in IPEX syndrome is heterogeneous and can include eczematous, psoriasiform, and ichthyosiform lesions, intermittent urticaria, alopecia universalis, onychodystrophy and pemphigoid lesions.
Currently, allogeneic HSCT is the best treatment option and should be performed before organ damage develops. Long-term follow-up reports a 10-year survival of 72,8% after HSCT (146). Immunosuppressive therapy is usually administrated after transplantation. Cyclosporine A, sirolimus and tacrolimus, or steroids are the most used agents. Rapamycin demonstrated to restore Treg cell function in IPEX syndrome, improving their suppression ability (147). It was demonstrated that immunosuppressive therapies alone do not impact the disease progression, and are associated with reduced life expectancy (148).
Recently, for the first time an IPEX patient with diffuse eczema was successfully treated with dupilumab. In this case, patient's dermatitis and itching persisted without improvement despite the HSCT and immune suppressive drugs (149).
Human T cells generated by viral transduction of a transcription unit encoding FOXP3, expressed a regulatory T phenotype in vitro (150) and could represent a novel therapeutic approach to modulate immune responses in the setting of allergy, autoimmunity, and immunodeficiencies. Initial trials of Treg-based cell therapy for IPEX syndrome are already tested in vitro and in animal models with promising results, but limitations are mainly related to the lifespan of the CD4 + T cells expressing wild-type FOXP3 (151).
5. Discussion
In the last decade, the rapid evolution of knowledge in the diagnosis and treatment of IEIs and the recognition of atopic disorders as a frequent feature have improved our knowledge of IEIs-A. Chronic skin disease is one of the main clinical manifestations in IEIs-As, and may manifest with eczema, erythroderma and urticaria. In particular, eczema is the most frequent manifestation and it is reported in 13%–22% of IEIs patients (152, 153).
In most cases, the skin involvement is similar to that found in non-immunocompromised patients, but when observed in IEIs-As, it tends to have an earlier onset, great severity, and possible complications such as infection. Another features of eczema in patients with IEIs-A is their unresponsiveness to conventional treatment that traditionally includes moisturizers, topical corticosteroids, and calcineurin inhibitors (152).
Figure 1 summarizes the warning signs that may guide to the diagnosis of IEIs-A.
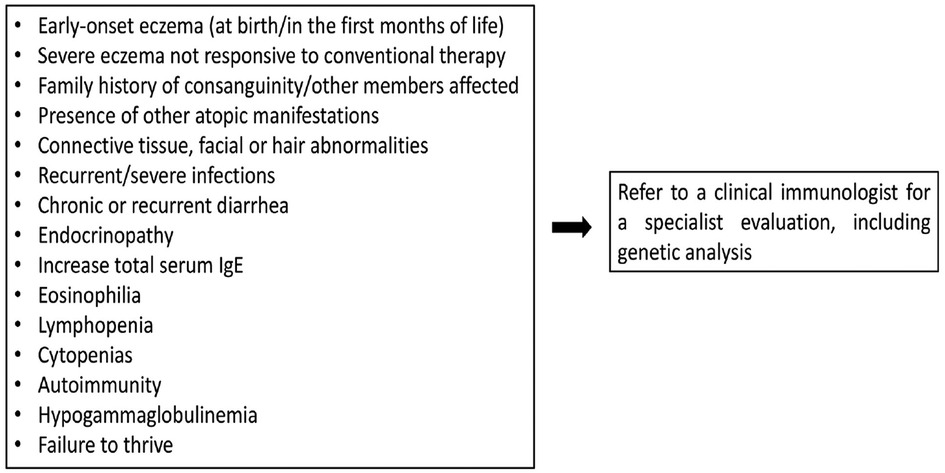
Figure 1. Warning signs for IEIs-A.
Continual improvements and accessibility of genetic analysis have helped to identify new IEIs-A diseases and to detect the intracellular pathway involved, allowing the possibility of precision therapy (154) (Figure 2). The goal of the tailored therapy is to use therapeutic agents to modulate dysfunctional pathways (155). Clinical evalutations to consider before starting biologic drugs are summarized in Figure 3.
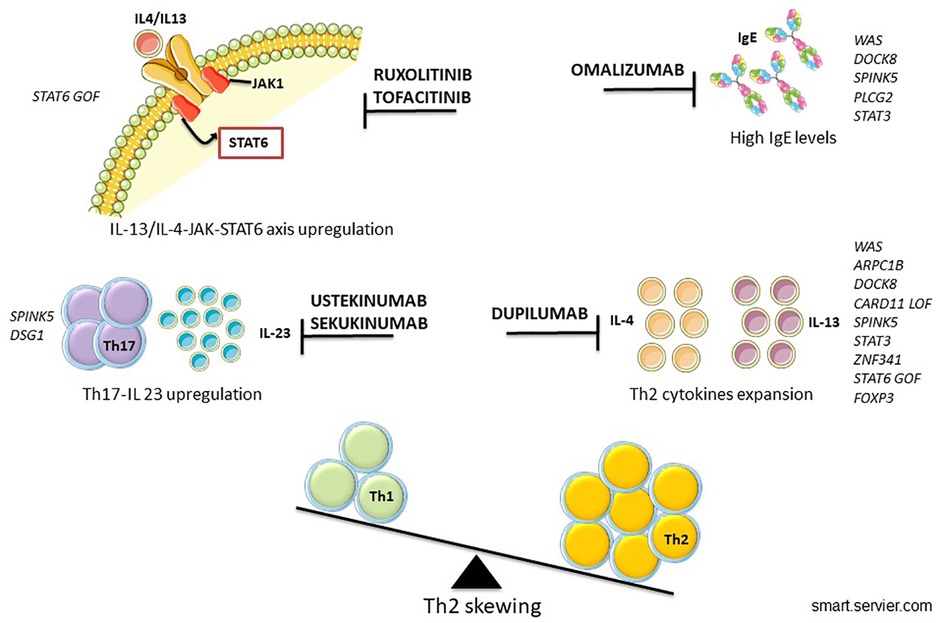
Figure 2. Proposed biologics in IEIs-A with skin involvement.

Figure 3. When to candidate for biologic treatment?
Recently, biologics became an interesting and promising option to treat refractory AD. Since 2020 dupilumab represents the first option among systemic therapies, and many randomized clinical trials have proven its safety and efficacy in both children and adolescents with AD (155–158).
Dupilumab is the most studied biological drug so far. It binds IL-4Rα inhibiting the IL-13/IL-4/STAT6 axis that includes cytokines with a crucial role in the pathogenesis of AD. In the context of IEIs-A, dupilumab has been used in many models with a Th2-skewed immune response with successful results (128). An increasing number of severe eczema treated with dupilumab IEIs-A patients are being reported in the literature (120, 159–161) and a recovery of Th1 polarization after its use is described in many cases (120).
The major advantage in treating immunocompromised patients with dupilumab is its safety profile as it does not cause further immunosuppression (162). In addition, it may be used as a bridge treatment in patients waiting for HSCT, in order to control skin infectious and inflammatory complications.
Omalizumab is currently approved for allergic asthma and chronic spontaneous urticaria. Its use in severe pediatric AD has been tested in a randomized clinical trial concluding that omalizumab is a treatment option for difficult-to-manage severe eczema in children with atopy (163). Omalizumab application in IEIs-A is limited to case reports or single clinical experience in particular in HIES with concomitant respiratory manifestations, but this application is still debated (63, 163–166).
In different forms of congenital ichthyosis the use of anti IL-17 A antibody and an IL-12/IL-23 antagonist have been proposed with promising results, based on an IL-17–skewed inflammatory signature revealed in these patients (86, 87).
In last years, many drugs involving different pathways are being studied to manage moderate and severe AD. All of the conventional treatments (steroid, cyclosporine, tacrolimus and dupilumab) inhibit the IL-13/IL-4-JAK-STAT6/STAT3 axis and the subsequent production of IL-13/IL-4 cytokines. Therefore, targeting this pathway would be a promising strategy also to develop new biologics for AD (167).
The rationale for the use of JAK inhibitors in AD is its role in controlling the transduction of the JAK-STAT signaling for Th2 cytokines. In the context of IEIs-A its use should be evaluated in selected cases where the axis is upregulated (167).
Clinical trials of new biologics for AD, already used in other diseases, include those targeting IL-13, IL-31RA, IL-33, OX40 and IL-22 (168). Their mechanism and phase of study are summarized in Table 2.
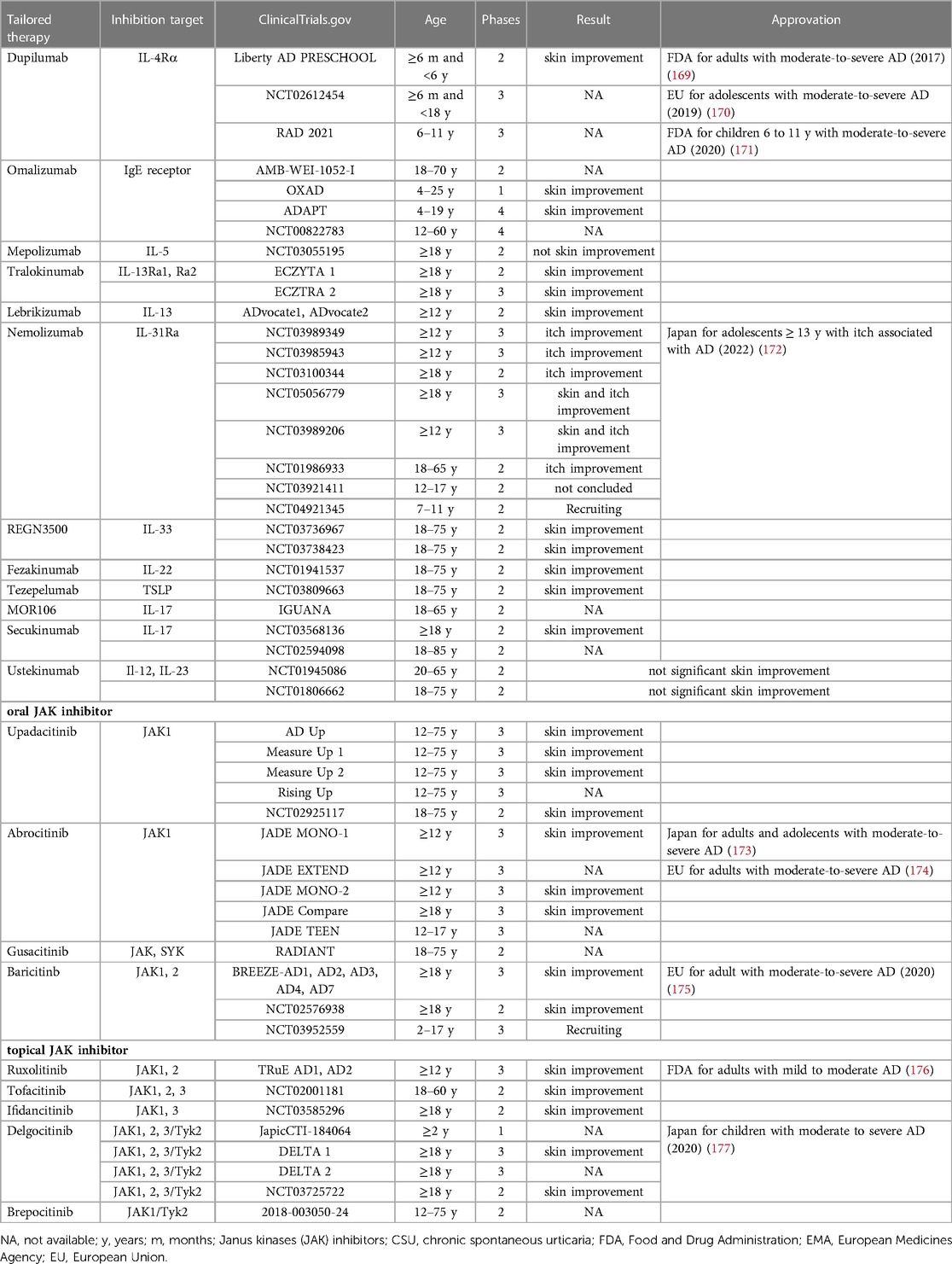
Table 2. Biological therapies for AD.
Targeted therapy advantages are related with its high specificity for one or few molecules, and therefore with its low toxicity compared to other systemic therapy for AD. This approach could be translated in IEIs-A with skin involvement in which such pathways are also affected. IEIs-A include a wide variety of diseases with different severity and prognosis and HSCT is the therapy of choice in a large number of these disorders, for which this treatment is potentially curative. In IEIs-A with immunodeficiency requiring HSCT, the use of biologics should not delay timing of transplantation. Indeed, biologics have proved to be effective in modulating an altered pathway and relieving symptoms but do not represnet a definitive therapy in immunodeficiencies. Rare diseases such as IEIs are inherently difficult to study in well-controlled clinical trials and therefore need a multidisciplinary management involving clinical immunologists and dermatologists to perform correct diagnosis and appropriate therapy in order to improve patient outcomes.
6. Conclusions
IEIs management is challenging but still affordable when a prompt diagnosis and an appropriate treatment are established. A diagnostic delay of IEIs is historically reported due to the variability of clinical phenotype and their rarity. However, the increasing availability of NGS technology together with recent research advances in IEIs and IEIs-A have improved the early diagnosis and optimized the treatment of these conditions. The speed, accuracy, and sensitivity of molecular analysis is crucial in the era of precision medicine based on a person's disease-driving molecular alterations. Biologics have the great advantage to act on a targeted component of immune system and they are becoming increasingly effective and safe for the therapeutic approach of many skin diseases.
There is an essential lack of knowledge about the efficacy of biologics in IEIs-A and only limited case reports describing their use in clinical practice are available. Long-term follow-up studies need to assess the safety and persistence of efficacy of each biologic.
More and larger international multicenter studies in this special population are necessary to evaluate the clinical profile of new drugs and to identify biological markers which will help to select patients who may benefit from tailored interventions.
Author contributions
PZ and CG conceptualized the review idea. PZ, CG and NC did the bibliography research and wrote the manuscript. PZ supervised the literature research and the manuscript redaction. LP, VS, DA, ECM, GAR, BR, AF, CC, AD, MEH contributed to the bibliography research and to a critical revision of the manuscript. PZ and CG supervised the manuscript revision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported also by the Italian Ministry of Health with “Current Research funds”.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AD, atopic dermatitis; AR, autosomal recessive; ADGRE2, adhesion protein-coupled receptor E2; Arp2/3, actin-related protein 2/3 complex; ARPC1B, actin-related protein 2/3 complex subunit 1B; BCL10, B cell CLL/lymphoma 10; CADINS, CARD11-associated atopy with dominant interference of NF-kB signaling; CAPE syndrome, CARD14-associated papulosquamous eruption syndrome; CARD, caspase recruitment domain; CBM complex, CARD-BCL10-MALT1 complex; CDSN, corneodesmosin; DOCK8, dedicator of cytokinesis 8; DSG1, desmoglein 1; FLG, filament-aggregating protein; FOXP3, forkhead box P3; GOF, gain of function; GvHD, graft-versus-host disease; HIES, hyper-immunoglobulin E syndromes; HSCT, hematopoietic stem cell transplantation; IEIs, inborn errors of immunity; IEIs-A, inborn errors of immunity with associated atopy; IPEX syndrome, immune dysregulation, polyendocrinopathy and enteropathy; IVIG, intravenous immunoglobulin; LDS, Loeys–Dietz syndrome; LEKTI, lympho-epithelial kazal-type-related inhibitor; LOF, loss of function; MALT1, mucosa-associated lymphoid tissue lymphoma translocation protein 1; MAPK or MAP kinase, mitogen-activated protein kinase; NGS, next-generation sequencing; NS, Comèl-Netherton syndrome; NS-ILC, typical ichthyosis linearis circumflexa Netherton syndrome; NS-SE, scaly erythroderma Netherton syndrome; Os, Omenn syndrome; OS, overall survival; PADs, primary atopic disorders; PGM3, phosphoglucomutase 3; PLCG2, phospholipase C gamma 2; PLTEID, platelet abnormalities with eosinophilia and immunomediated inflammatory disease; PSS, peeling skin syndrome; SAM, multiple allergies and metabolic wasting; SPINK5, inhibitor Kazal-type 5; STAT3, transducer and activator of transcription 3; TGFβ, transforming growth factor β; TYK2, tyrosine kinase 2; WAS, Wiskott-Aldrich syndrome; ZNF431, zinc finger protein 431.
References
1. Vaseghi-Shanjani M, Smith KL, Sara RJ, Modi BP, Branch A, Sharma M, et al. Inborn errors of immunity manifesting as atopic disorders. J Allergy Clin Immunol. (2021) 148:1130–9. doi: 10.1016/j.jaci.2021.08.008
2. Lyons JJ, Milner JD. Primary atopic disorders. J Exp Med. (2018) 215:1009–22. doi: 10.1084/jem.20172306
3. Candotti F. Clinical manifestations and pathophysiological mechanisms of the wiskott-aldrich syndrome. J Clin Immunol. (2018) 38:13–27. doi: 10.1007/s10875-017-0453-z
4. O’Connell AE, Volpi S, Dobbs K, Fiorini C, Tsitsikov E, de Boer H, et al. Next generation sequencing reveals skewing of the T and B cell receptor repertoires in patients with wiskott–aldrich syndrome. Front Immunol. (2014) 5:340. doi: 10.3389/fimmu.2014.00340
5. Du HQ, Zhang X, An YF, Ding Y, Zhao XD. Effects of W iskott–A ldrich syndrome protein deficiency on IL-10-producing regulatory B cells in humans and mice. Scand J Immunol. (2015) 81:483–93. doi: 10.1111/sji.12282
6. Rivers E, Thrasher AJ. Wiskott-Aldrich syndrome protein: emerging mechanisms in immunity. Eur J Immunol. (2017) 47:1857–66. doi: 10.1002/eji.201646715
7. Herman KE, Yoshida T, Hughson A, Grier A, Gill SR, Beck LA, et al. IL-17-Dependent dysregulated cutaneous immune homeostasis in the absence of the wiskott–aldrich syndrome protein. Front Immunol. (2022) 13:817427. doi: 10.3389/fimmu.2022.817427
8. Vasanna SH, Pereda MA, Dalal J. Clinical features, cancer biology, transplant approach and other integrated management strategies for wiskott–aldrich syndrome. J Multidiscip Healthc. (2021) 14:3497. doi: 10.2147/JMDH.S295386
9. Brigida I, Scaramuzza S, Lazarevic D, Cittaro D, Ferrua F, Leonardelli L, et al. A novel genomic inversion in wiskott-aldrich–associated autoinflammation. J Allergy Clin Immunol. (2016) 138:619–22. doi: 10.1016/j.jaci.2016.03.007
10. Gupta P, Subburaj K, Jindal A, Rawat A, Rikhi R, De D, et al. Atypical wiskott–aldrich syndrome without thrombocytopenia partially responding to omalizumab therapy. Clin Exp Dermatol. (2022) 47:1013–6. doi: 10.1111/ced.15119
11. Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinstitutional survey of the wiskott-aldrich syndrome. J Pediatr. (1994) 125:876–85. doi: 10.1016/S0022-3476(05)82002-5
12. Chandra S, Bronicki L, Nagaraj CB, Zhang K. WAS-related disorders. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle (2016).
13. Cavazzana M, Thrasher A. Gene therapy for whiskott–aldrich syndrome: the latest news. Clin Transl Med. (2022) 12:e815. doi: 10.1002/ctm2.815
14. Burroughs LM, Petrovic A, Brazauskas R, Liu X, Griffith LM, Ochs HD, et al. Excellent outcomes following hematopoietic cell transplantation for wiskott-aldrich syndrome: a PIDTC report. Blood. (2020) 135:2094–105. doi: 10.1182/blood.2019002939
15. Albert MH, Slatter MA, Gennery AR, Güngör T, Bakunina K, Markovitch B, et al. Hematopoietic stem cell transplantation for wiskott-aldrich syndrome: an EBMT inborn errors working party analysis. Blood. J Am Soc Hematol. (2022) 139:2066–79. doi: 10.1182/blood.2021014687
16. Rivers E, Worth A, Thrasher AJ, Burns SO. How I manage patients with wiskott aldrich syndrome. Br J Haematol. (2019) 185:647–55. doi: 10.1111/bjh.15831
17. Morris EC. Allogeneic hematopoietic stem cell transplantation in adults with primary immunodeficiency. In: Hematology 2014, the American society of hematology education program book. (2020) 2020(1):649–60. doi: 10.1182/hematology.2020000152
18. Consiglieri G, Ferrua F, Aiuti A, Cicalese MP. A case of two adult brothers with wiskott-aldrich syndrome, one treated with gene therapy and one with HLA-identical hematopoietic stem cell transplantation. J Clin Immunol. (2022) 42:421–5. doi: 10.1007/s10875-021-01157-6
19. Ferrua F, Cicalese MP, Galimberti S, Giannelli S, Dionisio F, Barzaghi F, et al. Lentiviral haemopoietic stem/progenitor cell gene therapy for treatment of wiskott-aldrich syndrome: interim results of a non-randomised, open-label, phase 1/2 clinical study. Lancet Haematol. (2019) 6:e239–253. doi: 10.1016/S2352-3026(19)30021-3
20. Cinicola BL, Corrente S, Castagnoli R, Lougaris V, Giardino G, Leonardi L, et al. Primary atopic disorders and chronic skin disease. Pediatr Allergy Immunol. (2022) 33:65–8. doi: 10.1111/pai.13633
21. Chiriaco M, Ursu GM, Amodio D, Cotugno N, Volpi S, Berardinelli F, et al. Radiosensitivity in patients affected by ARPC1B deficiency: a new disease trait? Front Immunol. (2022) 13:919237. doi: 10.3389/fimmu.2022.919237
22. Volpi S, Cicalese MP, Tuijnenburg P, Tool ATJ, Cuadrado E, Abu-Halaweh M, et al. A combined immunodeficiency with severe infections, inflammation, and allergy caused by ARPC1B deficiency. J Allergy Clin Immunol. (2019) 143:2296–9. doi: 10.1016/j.jaci.2019.02.003
23. Giardino S, Volpi S, Lucioni F, Caorsi R, Schneiderman J, Lang A, et al. Hematopoietic stem cell transplantation in ARPC1B deficiency. J Clin Immunol. (2022) 42:1535–44. doi: 10.1007/s10875-022-01305-6
24. Biggs CM, Keles S, Chatila TA. DOCK8 Deficiency: insights into pathophysiology, clinical features and management. Clin Immunol. (2017) 181:75–82. doi: 10.1016/j.clim.2017.06.003
25. Ollech A, Mashiah J, Lev A, Simon AJ, Somech R, Adam E, et al. Treatment options for DOCK8 deficiency-related severe dermatitis. J Dermatol. (2021) 48:1386–93. doi: 10.1111/1346-8138.15955
26. Gomes N, Miranda J, Lopes S, Carneiro-Leão L, Costa JT, Baudrier T, et al. Omalizumab in the treatment of hyper-IgE syndrome: 2 case reports. J Investig Allergol Clin Immunol. (2020) 30(3):191–2. doi: 10.18176/jiaci.0469
27. Aydin SE, Kilic SS, Aytekin C, Kumar A, Porras O, Kainulainen L, et al. DOCK8 Deficiency: clinical and immunological phenotype and treatment options-a review of 136 patients. J Clin Immunol. (2015) 35:189–98. doi: 10.1007/s10875-014-0126-0
28. Aydin SE, Freeman AF, Al-Herz W, Al-Mousa HA, Arnaout RK, Aydin RC, et al. Hematopoietic stem cell transplantation as treatment for patients with DOCK8 deficiency. J Allergy Clin Immunol Pract. (2019) 7:848–55. doi: 10.1016/j.jaip.2018.10.035
29. Lu HY, Bauman BM, Arjunaraja S, Dorjbal B, Milner JD, Snow AL, et al. The CBM-opathies—a rapidly expanding spectrum of human inborn errors of immunity caused by mutations in the CARD11-BCL10-MALT1 complex. Front Immunol. (2018) 9:2078. doi: 10.3389/fimmu.2018.02078
30. Arjunaraja S, Malinverni C, Sukumar G, Quanchard J, Lott N, Dalgard C, et al. Enhanced survival of BENTA patient B cells is dependent on MALT1 protease activity. J Immunol. (2017) 198:59.7. doi: 10.4049/jimmunol.198.Supp.59.7
31. Rozmus J, McDonald R, Fung SY, Del Bel KL, Roden J, Senger C, et al. Successful clinical treatment and functional immunological normalization of human MALT1 deficiency following hematopoietic stem cell transplantation. Clin Immunol. (2016) 168:1–5. doi: 10.1016/j.clim.2016.04.011
32. Punwani D, Wang H, Chan AY, Cowan MJ, Mallott J, Sunderam U, et al. Combined immunodeficiency due to MALT1 mutations, treated by hematopoietic cell transplantation. J Clin Immunol. (2015) 35:135–46. doi: 10.1007/s10875-014-0125-1
33. Jabara HH, Ohsumi T, Chou J, Massaad MJ, Benson H, Megarbane A, et al. A homozygous mucosa-associated lymphoid tissue 1 (MALT1) mutation in a family with combined immunodeficiency. J Allergy Clin Immunol. (2013) 132:151–8. doi: 10.1016/j.jaci.2013.04.047
34. Sefer AP, Abolhassani H, Ober F, Kayaoglu B, Bilgic Eltan S, Kara A, et al. Expanding the clinical and immunological phenotypes and natural history of MALT1 deficiency. J Clin Immunol. (2022) 42:634–52. doi: 10.1007/s10875-021-01191-4
35. Hutcherson SM, Bedsaul JR, Pomerantz JL. Pathway-specific defects in T, B, and NK cells and age-dependent development of high IgE in mice heterozygous for a CADINS-associated dominant negative CARD11 allele. J Immunol. (2021) 207:1150–64. doi: 10.4049/jimmunol.2001233
36. Urdinez L, Erra L, Palma AM, Mercogliano MF, Fernandez JB, Prieto E, et al. Expanding spectrum, intrafamilial diversity, and therapeutic challenges from 15 patients with heterozygous CARD11-associated diseases: a single center experience. Front Immunol. (2022) 13:1020927. doi: 10.3389/fimmu.2022.1020927
37. Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. (2014) 40:692–705. doi: 10.1016/j.immuni.2014.04.007
38. Zwol AV, Moll HA, Fetter WPF, Elburg RMV. Glutamine-enriched enteral nutrition in very low birthweight infants and allergic and infectious diseases at 6 years of age. Paediatr Perinat Epidemiol. (2011) 25:60–6. doi: 10.1111/j.1365-3016.2010.01173.x
39. Ma CA, Stinson JR, Zhang Y, Abbott JK, Weinreich MA, Hauk PJ, et al. Germline hypomorphic CARD11 mutations in severe atopic disease. Nat Genet. (2017) 49:1192–201. doi: 10.1038/ng.3898
40. Diaz-Cabrera N, Bauman B, Iro M, Dabbah G, Molho-Pessach V, Zlotogorski A, et al. Moderate to severe CARD11 loss of function-associated atopic dermatitis treated with biologic modifiers. J Clin Immunol. (2021) 46(7):1334–5. doi: 10.1007/s10875-021-01001-x
41. Charvet E, Bourrat E, Hickman G, Donadieu J, Bellanné-Chantelot C, Jachiet M, et al. Efficacy of dupilumab for controlling severe atopic dermatitis with dominant-negative CARD11 variant. Clin Exp Dermatol. (2021) 46:1334–5. doi: 10.1111/ced.14686
42. Pietzsch L, Körholz J, Boschann F, Sergon M, Dorjbal B, Yee D, et al. Hyper-IgE and carcinoma in CADINS disease. Front Immunol. (2022) 13:878989. doi: 10.3389/fimmu.2022.878989
43. Jordan CT, Cao L, Roberson EDO, Duan S, Helms CA, Nair RP, et al. Rare and common variants in CARD14, encoding an epidermal regulator of NF-kappaB, in psoriasis. Am J Hum Genet. (2012) 90:796–808. doi: 10.1016/j.ajhg.2012.03.013
44. Fuchs-Telem D, Sarig O, van Steensel MAM, Isakov O, Israeli S, Nousbeck J, et al. Familial pityriasis rubra pilaris is caused by mutations in CARD14. Am J Hum Genet. (2012) 91:163–70. doi: 10.1016/j.ajhg.2012.05.010
45. Frare CP, Blumstein AJ, Paller AS, Pieretti L, Choate KA, Bowcock AM, et al. CARD14-associated Papulosquamous eruption (CAPE) in pediatric patients: three additional cases and review of the literature. Pediatr Dermatol. (2021) 38:1237–42. doi: 10.1111/pde.14779
46. Peled A, Sarig O, Sun G, Samuelov L, Ma CA, Zhang Y, et al. Loss-of-function mutations in caspase recruitment domain-containing protein 14 (CARD14) are associated with a severe variant of atopic dermatitis. J Allergy Clin Immunol. (2019) 143:173–81. doi: 10.1016/j.jaci.2018.09.002
47. Jordan CT, Cao L, Roberson EDO, Pierson KC, Yang CF, Joyce CE, et al. PSORS2 Is due to mutations in CARD14. Am J Hum Genet. (2012) 90:784–95. doi: 10.1016/j.ajhg.2012.03.012
48. Craiglow BG, Boyden LM, Hu R, Virtanen M, Su J, Rodriguez G, et al. CARD14-associated Papulosquamous eruption: a spectrum including features of psoriasis and pityriasis rubra pilaris. J Am Acad Dermatol. (2018) 79:487–94. doi: 10.1016/j.jaad.2018.02.034
49. Kiszewski AE, De Almeida Jr HL. Successful treatment with ustekinumab in CARD14-associated papulosquamous eruption in a Brazilian child. Dermatol Ther. (2022) 35(12):e15939. doi: 10.1111/dth.15939
50. Eytan O, Sarig O, Sprecher E, van Steensel MAM. Clinical response to ustekinumab in familial pityriasis rubra pilaris caused by a novel mutation in CARD14. Br J Dermatol. (2014) 171:420–2. doi: 10.1111/bjd.12952
51. Benson JM, Peritt D, Scallon BJ, Heavner GA, Shealy DJ, Giles-Komar JM, et al. Discovery and mechanism of ustekinumab: a human monoclonal antibody targeting interleukin-12 and interleukin-23 for treatment of immune-mediated disorders. In MAbs. (2011) 3(6):535–45. doi: 10.4161/mabs.3.6.17815
52. Orphanet. Comel Netherton Syndrome (2008). Available at: www.orpha.net (2008 October).
53. Chavanas S, Bodemer C, Rochat A, Hamel-Teillac D, Ali M, Irvine AD, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause netherton syndrome. Nat Genet. (2000) 25:141–2. doi: 10.1038/75977
54. Pruszkowski A, Bodemer C, Fraitag S, Teillac-Hamel D, Amoric JC, de Prost Y. Neonatal and infantile erythrodermas: a retrospective study of 51 patients. Arch Dermatol. (2000) 136:875–80. doi: 10.1001/archderm.136.7.875
55. Barbati F, Giovannini M, Oranges T, Lodi L, Barni S, Novembre E, et al. Netherton syndrome in children: management and future perspectives. Front Pediatr. (2021) 9:645259. doi: 10.3389/fped.2021.645259
56. Berna R, Mitra N, Lou C, Wan J, Hoffstad O, Wubbenhorst B, et al. Thymic stromal lymphopoietin and IL7R variants are associated with persistent atopic dermatitis. J Invest Dermatol. (2021) 141:446. doi: 10.1016/j.jid.2020.05.119
57. Volc S, Maier L, Gritsch A, Aichelburg MC, Volc-Platzer B. Successful treatment of netherton syndrome with ustekinumab in a 15-year-old girl. Br J Dermatol. (2020) 183:165–7. doi: 10.1111/bjd.18892
58. Barbieux C, Des Claustres MB, Fahrner M, Petrova E, Tsoi LC, Gouin O, et al. Netherton syndrome subtypes share IL-17/IL-36 signature with distinct IFN-α and allergic responses. J Allergy Clin Immunol. (2022) 149:1358–72. doi: 10.1016/j.jaci.2021.08.024
59. Small AM, Cordoro KM. Netherton syndrome mimicking pustular psoriasis: clinical implications and response to intravenous immunoglobulin. Pediatr Dermatol. (2016) 33:e222–223. doi: 10.1111/pde.12856
60. Zhang Z, Pan C, Wei R, Li H, Yang Y, Chen J, et al. Netherton syndrome caused by compound heterozygous mutation, c. 80A > G mutation in SPINK5 and large-sized genomic deletion mutation, and successful treatment of intravenous immunoglobulin. Mol Genet Genomic Med. (2021) 9:e1600. doi: 10.1002/mgg3.1600
61. Nouwen AEM, Schappin R, Nguyen NT, Ragamin A, Bygum A, Bodemer C, et al. Outcomes of systemic treatment in children and adults with netherton syndrome; a systematic review. Front Immunol. (2022) 13:864449. doi: 10.3389/fimmu.2022.864449
62. Renner ED, Hartl D, Rylaarsdam S, Young ML, Monaco-Shawver L, Kleiner G, et al. Comel-Netherton syndrome defined as primary immunodeficiency. J Allergy Clin Immunol. (2009) 124:536–43. doi: 10.1016/j.jaci.2009.06.009
63. Wang HH, Li YC, Huang YC. Efficacy of omalizumab in patients with atopic dermatitis: a systematic review and meta-analysis. J Allergy Clin Immunol. (2016) 138:1719–22. doi: 10.1016/j.jaci.2016.05.038
64. Kaveri SV, Maddur MS, Hegde P, Lacroix-Desmazes S, Bayry J. Intravenous immunoglobulins in immunodeficiencies: more than mere replacement therapy. Clin Exp Immunol. (2011) 164:2–5. doi: 10.1111/j.1365-2249.2011.04387.x
65. Ragamin A, Nouwen AEM, Dalm VASH, van Mierlo MMF, Lincke CR, Pasmans SGMA. Treatment experiences with intravenous immunoglobulins, ixekizumab, dupilumab, and anakinra in netherton syndrome: a case series. Dermatology. (2022) 239(1):72–80. doi: 10.1159/000525987
66. Süßmuth K, Traupe H, Loser K, Ständer S, Kessel C, Wittkowski H, et al. Response to dupilumab in two children with netherton syndrome: improvement of pruritus and scaling. J Eur Acad Dermatol Venereol. (2021) 35:e152–155. doi: 10.1111/jdv.16883
67. Liddle J, Beneton V, Benson M, Bingham R, Bouillot A, Boullay AB, et al. A potent and selective kallikrein-5 inhibitor delivers high pharmacological activity in skin from patients with netherton syndrome. J Invest Dermatol. (2021) 141:2272–9. doi: 10.1016/j.jid.2021.01.029
68. Di WL, Larcher F, Semenova E, Talbot GE, Harper JI, Del Rio M, et al. Ex-vivo gene therapy restores LEKTI activity and corrects the architecture of netherton syndrome-derived skin grafts. Mol Ther. (2011) 19:408–16. doi: 10.1038/mt.2010.201
69. Di WL, Mellerio JE, Bernadis C, Harper J, Abdul-Wahab A, Ghani S, et al. Phase I study protocol for ex vivo lentiviral gene therapy for the inherited skin disease, netherton syndrome. Hum Gene Ther Clin Dev. (2013) 24:182–90. doi: 10.1089/humc.2013.195
70. ClinicalTrials.gov. NCT01545323. Available at: https://clinicaltrials.gov/ct2/show/NCT01545323
71. Murase C, Takeichi T, Taki T, Yoshikawa T, Suzuki A, Ogi T, et al. Successful dupilumab treatment for ichthyotic and atopic features of netherton syndrome. J Dermatol Sci. (2021) 102:126–9. doi: 10.1016/j.jdermsci.2021.03.003
72. Steuer AB, Cohen DE. Treatment of netherton syndrome with dupilumab. JAMA Dermatol. (2020) 156:350–1. doi: 10.1001/jamadermatol.2019.4608
73. Andreasen TH, Karstensen HG, Duno M, Lei U, Zachariae C, Thyssen JP. Successful treatment with dupilumab of an adult with netherton syndrome. Clin Exp Dermatol. (2020) 45:915–7. doi: 10.1111/ced.14317
74. ClinicalTrials.gov. NCT04244006. Available at: https://clinicaltrials.gov/ct2/show/NCT04244006
75. Yalcin AD. A case of netherton syndrome: successful treatment with omalizumab and pulse prednisolone and its effects on cytokines and immunoglobulin levels. Immunopharmacol Immunotoxicol. (2016) 38:162–6. doi: 10.3109/08923973.2015.1115518
76. Roda Â, Mendonça-Sanches M, Travassos AR, de-Almeida LS, Metze D. Infliximab therapy for netherton syndrome: a case report. JAAD Case Rep. (2017) 3:550. doi: 10.1016/j.jdcr.2017.07.019
77. Luchsinger I, Knöpfel N, Theiler M, Des Claustres MB, Barbieux C, Schwieger-Briel A, et al. Secukinumab therapy for netherton syndrome. JAMA Dermatol. (2020) 156:907–11. doi: 10.1001/jamadermatol.2020.1019
78. Blanchard SK, Prose NS. Successful use of secukinumab in netherton syndrome. JAAD Case Rep. (2020) 6:577–8. doi: 10.1016/j.jdcr.2020.04.025
79. Barbieux C, Des Claustres MB, De la Brassinne M, Bricteux G, Bagot M, Bourrat E, et al. Duality of netherton syndrome manifestations and response to ixekizumab. J Am Acad Dermatol. (2021) 84:1476–80. doi: 10.1016/j.jaad.2020.07.054
80. Czarnowicki T, He H, Krueger JG, Guttman-Yassky E. Atopic dermatitis endotypes and implications for targeted therapeutics. J Allergy Clin Immunol. (2019) 143:1–11. doi: 10.1016/j.jaci.2018.10.032
81. Astolfi A, Cipriani F, Messelodi D, De Luca M, Indio V, Di Chiara C, et al. Filaggrin loss-of-function mutations are risk factors for severe food allergy in children with atopic dermatitis. J Clin Med. (2021) 10:233. doi: 10.3390/jcm10020233
82. Otsuka A, Doi H, Egawa G, Maekawa A, Fujita T, Nakamizo S, et al. Possible new therapeutic strategy to regulate atopic dermatitis through upregulating filaggrin expression. J Allergy Clin Immunol. (2014) 133:139–46. doi: 10.1016/j.jaci.2013.07.027
83. Stout TE, McFarland T, Mitchell JC, Appukuttan B, Stout JT. Recombinant filaggrin is internalized and processed to correct filaggrin deficiency. J Invest Dermatol. (2014) 134:423–9. doi: 10.1038/jid.2013.284
84. Czarnowicki T, Malajian D, Khattri S, Correa da Rosa J, Dutt R, Finney R, et al. Petrolatum: barrier repair and antimicrobial responses underlying this “inert” moisturizer. J Allergy Clin Immunol. (2016) 137:1091–102. doi: 10.1016/j.jaci.2015.08.013
85. Samuelov L, Sarig O, Harmon RM, Rapaport D, Ishida-Yamamoto A, Isakov O, et al. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet. (2013) 45:1244–8. doi: 10.1038/ng.2739
86. Frommherz L, Schempp CM, Has C. Secukinumab for the treatment of SAM syndrome associated with desmoglein-1 deficiency. Br J Dermatol. (2020) 184(4):770–2. doi: 10.1111/bjd.19684
87. Godsel LM, Roth-Carter QR, Koetsier JL, Tsoi LC, Huffine AL, Broussard JA, et al. Translational implications of Th17-skewed inflammation due to genetic deficiency of a cadherin stress sensor. J Clin Invest. (2022) 132:e144363. doi: 10.1172/JCI144363
88. Lee UH, Kim BE, Kim DJ, Cho YG, Ye YM, Leung DYM. Atopic dermatitis is associated with reduced corneodesmosin expression: role of cytokine modulation and effects on viral penetration. Br J Dermatol. (2017) 176:537–40. doi: 10.1111/bjd.15010
89. Galli L, Venturini E, Bassi A, Gattinara GC, Chiappini E, Defilippi C, et al. Common community-acquired bacterial skin and soft-tissue infections in children: an intersociety consensus on impetigo, abscess, and cellulitis treatment. Clin Ther. (2019) 41:532–51. doi: 10.1016/j.clinthera.2019.01.010
90. Valentin F, Wiegmann H, Tarinski T, Nikolenko H, Traupe H, Liebau E, et al. Development of a pathogenesis-based therapy for peeling skin syndrome type 1. Br J Dermatol. (2021) 184:1123–31. doi: 10.1111/bjd.19546
91. Naranjo AN, Bandara G, Bai Y, Smelkinson MG, Tobío A, Komarow HD, et al. Critical signaling events in the mechanoactivation of human mast cells through p. C492Y-ADGRE2. J Invest Dermatol. (2020) 140:2210–20. doi: 10.1016/j.jid.2020.03.936
92. Milner JD. PLAID: a syndrome of complex patterns of disease and unique phenotypes. J Clin Immunol. (2015) 35:527–30. doi: 10.1007/s10875-015-0177-x
93. Martín-Nalda A, Fortuny C, Rey L, Bunney TD, Alsina L, Esteve-Solé A, et al. Severe autoinflammatory manifestations and antibody deficiency due to novel hypermorphic PLCG2 mutations. J Clin Immunol. (2020) 40:987–1000. doi: 10.1007/s10875-020-00794-7
94. Welzel T, Oefelein L, Holzer U, Müller A, Menden B, Haack TB, et al. Variant in the PLCG2 gene may cause a phenotypic overlap of APLAID/PLAID: case series and literature review. J Clin Med. (2022) 11:4369. doi: 10.3390/jcm11154369
95. García-García A, Arellano MB, Deyà-Martínez À, Blasco JL, Serrano M, Rym AVD, et al. Novel PGM3 compound heterozygous variants with IgE-related dermatitis, lymphopenia, without syndromic features. Pediatr Allergy Immunol. (2021) 32:566–75. doi: 10.1111/pai.13398
96. Zhang Y, Yu X, Ichikawa M, Lyons JJ, Datta S, Lamborn IT, et al. Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J Allergy Clin Immunol. (2014) 133:1400–9. doi: 10.1016/j.jaci.2014.02.013
97. Stray-Pedersen A, Backe PH, Sorte HS, Mørkrid L, Chokshi NY, Erichsen HC, et al. PGM3 Mutations cause a congenital disorder of glycosylation with severe immunodeficiency and skeletal dysplasia. Am J Hum Genet. (2014) 95:96–107. doi: 10.1016/j.ajhg.2014.05.007
98. Harms HK, Zimmer KP, Kurnik K, Bertele-Harms RM, Weidinger S, Reiter K. Oral mannose therapy persistently corrects the severe clinical symptoms and biochemical abnormalities of phosphomannose isomerase deficiency. Acta Paediatr. (2002) 91:1065–72. doi: 10.1111/j.1651-2227.2002.tb00101.x
99. ClinicalTrials.gov. NCT02511041. Available at: https://clinicaltrials.gov/ct2/show/NCT02511041
100. Nelson RW, Geha RS, McDonald DR. Inborn errors of immunity associated with atopy. Front Immunol. (2022) 13:860821. doi: 10.3389/fimmu.2022.860821
101. Gomez L, Le Deist F, Blanche S, Cavazzana-Calvo M, Griscelli C, Fischer A. Treatment of omenn syndrome by bone marrow transplantation. J Pediatr. (1995) 127:76–81. doi: 10.1016/S0022-3476(95)70260-1
102. Meyer-Bahlburg A, Haas JP, Haase R, Eschrich U, Wawer A, Frank L, et al. Treatment with cyclosporin A in a patient with omenn’s syndrome. Arch Dis Child. (2002) 87:231–3. doi: 10.1136/adc.87.3.231
103. de Saint-Basile G, Le Deist F, De Villartay JP, Cerf-Bensussan N, Journet O, Brousse N, et al. Restricted heterogeneity of T lymphocytes in combined immunodeficiency with hypereosinophilia (omenn's syndrome). J Clin Invest. (1991) 87:1352–9. doi: 10.1172/JCI115139
104. Villa A, Notarangelo LD, Roifman CM. Omenn syndrome: inflammation in leaky severe combined immunodeficiency. J Allergy Clin Immunol. (2008) 122:1082–6. doi: 10.1016/j.jaci.2008.09.037
105. Ahn T, Sheng G, Garcia-Lloret M, Butte M. A proposed targeted treatment for omenn syndrome A case report. J Clin Immunol. (2021) 41(Suppl 1):1–135. doi: 10.1007/s10875-021-01001-x
106. Capo V, Castiello MC, Fontana E, Penna S, Bosticardo M, Draghici E, et al. Efficacy of lentivirus-mediated gene therapy in an omenn syndrome recombination-activating gene 2 mouse model is not hindered by inflammation and immune dysregulation. J Allergy Clin Immunol. (2018) 142:928–41. doi: 10.1016/j.jaci.2017.11.015
107. Pike-Overzet K, Rodijk M, Ng YY, Baert MRM, Lagresle-Peyrou C, Schambach A, et al. Correction of murine Rag1 deficiency by self-inactivating lentiviral vector-mediated gene transfer. Leukemia. (2011) 25:1471–83. doi: 10.1038/leu.2011.106
108. Villa A, Capo V, Castiello MC. Innovative cell-based therapies and conditioning to cure RAG deficiency. Front Immunol. (2020) 11:607926. doi: 10.3389/fimmu.2020.607926
109. Garcia-Perez L, van Eggermond M, van Roon L, Vloemans SA, Cordes M, Schambach A, et al. Successful preclinical development of gene therapy for recombinase-activating gene-1-deficient SCID. Mol Ther Methods Clin Dev. (2020) 17:666–82. doi: 10.1016/j.omtm.2020.03.016
110. Tsilifis C, Freeman AF, Gennery AR. STAT3 hyper-IgE syndrome—an update and unanswered questions. J Clin Immunol. (2021) 41:864–80. doi: 10.1007/s10875-021-01051-1
111. Ponsford MJ, Klocperk A, Pulvirenti F, Dalm VASH, Milota T, Cinetto F, et al. Hyper-IgE in the allergy clinic—when is it primary immunodeficiency? Allergy. (2018) 73:2122–36. doi: 10.1111/all.13578
112. Chandesris MO, Melki I, Natividad A, Puel A, Fieschi C, Yun L, et al. Autosomal dominant STAT3 deficiency and hyper-IgE syndrome molecular, cellular, and clinical features from a French national survey. Medicine (Baltimore). (2012) 91:e1. doi: 10.1097/MD.0b013e31825f95b9
113. Nester TA, Wagnon AH, Reilly WF, Spitzer G, Kjeldsberg CR, Hill HR. Effects of allogeneic peripheral stem cell transplantation in a patient with job syndrome of hyperimmunoglobulinemia E and recurrent infections. Am J Med. (1998) 105:162–4. doi: 10.1016/S0002-9343(98)00200-9
114. Gennery AR, Flood TJ, Abinun M, Cant AJ. Bone marrow transplantation does not correct the hyper IgE syndrome. Bone Marrow Transplant. (2000) 25:1303–5. doi: 10.1038/sj.bmt.1702446
115. Yanagimachi M, Ohya T, Yokosuka T, Kajiwara R, Tanaka F, Goto H, et al. The potential and limits of hematopoietic stem cell transplantation for the treatment of autosomal dominant hyper-IgE syndrome. J Clin Immunol. (2016) 36:511–6. doi: 10.1007/s10875-016-0278-1
116. Oikonomopoulou C, Goussetis E. Autosomal dominant hyper-IgE syndrome: when hematopoietic stem cell transplantation should be considered? Pediatr Transplant. (2020) 24:e13699. doi: 10.1111/petr.13699
117. Harrison SC, Tsilifis C, Slatter MA, Nademi Z, Worth A, Veys P, et al. Hematopoietic stem cell transplantation resolves the immune deficit associated with STAT3-dominant-negative hyper-IgE syndrome. J Clin Immunol. (2021) 41:934–43. doi: 10.1007/s10875-021-00971-2
118. Sogkas G, Hirsch S, Jablonka A, Witte T, Schmidt RE, Atschekzei F. Dupilumab to treat severe atopic dermatitis in autosomal dominant hyper-IgE syndrome. Clin Immunol. (2020) 215:108452. doi: 10.1016/j.clim.2020.108452
119. Staudacher O, Krüger R, Kölsch U, Thee S, Gratopp A, Wahn V, et al. Relieving job: dupilumab in autosomal dominant STAT3 hyper-IgE syndrome. J Allergy Clin Immunol Pract. (2022) 10:349–51. doi: 10.1016/j.jaip.2021.08.042
120. Matucci-Cerinic C, Viglizzo G, Pastorino C, Corcione A, Prigione I, Bocca P, et al. Remission of eczema and recovery of Th1 polarization following treatment with dupilumab in STAT3 hyper IgE syndrome. Pediatr Allergy Immunol. (2022) 33:e13770. doi: 10.1111/pai.13770
121. Wang HJ, Yang TT, Lan CCE. Dupilumab treatment of eczema in a child with STAT3 hyper-immunoglobulin E syndrome. J Eur Acad Dermatol Venereol. (2022) 36:e367–369. doi: 10.1111/jdv.17889
122. Bard S, Paravisini A, Avilés-Izquierdo JA, Fernandez-Cruz E, Sánchez-Ramón S. Eczematous dermatitis in the setting of hyper-IgE syndrome successfully treated with omalizumab. Arch Dermatol. (2008) 144:1662–3. doi: 10.1001/archdermatol.2008.510
123. Alonso-Bello CD, Jiménez-Martínez M, Vargas-Camaño ME, Hierro-Orozco S, Ynga-Durand MA, Berrón-Ruiz L, et al. Partial and transient clinical response to omalizumab in IL-21-induced low STAT3-phosphorylation on hyper-IgE syndrome. Case Reports Immunol. (2019) 2019:6357256. doi: 10.1155/2019/6357256
124. Marcotte GV. Omalizumab therapy for hyper-IgE syndrome. J Allergy Clin Immunol. (2008) 121:S88. doi: 10.1016/j.jaci.2007.12.353
125. Lan J, Zhang Y, Song M, Cai S, Luo H, OuYang R, et al. Omalizumab for STAT3 hyper-IgE syndromes in adulthood: a case report and literature review. Front Med (Lausanne). (2022) 9:835257. doi: 10.3389/fmed.2022.835257
126. Papaioannou O, Karampitsakos T, Katsaras M, Sampsonas F, Tzouvelekis A. Clinical improvement in job syndrome following administration of co-trimoxazole, omalizumab and inhaled tobramycin. Adv Respir Med. (2021) 89:585–8. doi: 10.5603/ARM.a2021.0079
127. Cekic S, Huriyet H, Hortoglu M, Kasap N, Ozen A, Karakoc-Aydiner E, et al. Increased radiosensitivity and impaired DNA repair in patients with STAT3-LOF and ZNF341 deficiency, potentially contributing to malignant transformations. Clin Exp Immunol. (2022) 209:83–9. doi: 10.1093/cei/uxac041
128. Lévy R, Béziat V, Barbieux C, Puel A, Bourrat E, Casanova JL, et al. Efficacy of dupilumab for controlling severe atopic dermatitis in a patient with hyper-IgE syndrome. J Clin Immunol. (2020) 40:418–20. doi: 10.1007/s10875-020-00751-4
129. Kreins AY, Ciancanelli MJ, Okada S, Kong XF, Ramírez-Alejo N, Kilic SS, et al. Human TYK2 deficiency: mycobacterial and viral infections without hyper-IgE syndrome. J Exp Med. (2015) 212:1641–62. doi: 10.1084/jem.20140280
130. Wu P, Chen S, Wu B, Chen J, Lv G. A TYK2 gene mutation c. 2395G > A leads to TYK2 deficiency: a case report and literature review. Front Pediatr. (2020) 8:253. doi: 10.3389/fped.2020.00253
131. Bousfiha A, Moundir A, Tangye SG, Picard C, Jeddane L, Al-Herz W, et al. The 2022 update of IUIS phenotypical classification for human inborn errors of immunity. J Clin Immunol. (2022) 42(7):1508–20. doi: 10.1007/s10875-020-00758-x
132. Bieber T. Atopic dermatitis: an expanding therapeutic pipeline for a complex disease. Nat Rev Drug Discovery. (2022) 21:21–40. doi: 10.1038/s41573-021-00266-6
133. Minegishi Y. Hyper-IgE syndrome, 2021 update. Allergol Int. (2021) 70:407–14. doi: 10.1016/j.alit.2021.07.007
134. Kiwanuka KN, Kolawole EM, Mcleod JJA, Baker B, Paez PA, Zellner MP, et al. Stat5B is required for IgE-mediated mast cell function in vitro and in vivo. Cell Immunol. (2021) 364:104344. doi: 10.1016/j.cellimm.2021.104344
135. Karpathiou G, Papoudou-Bai A, Ferrand E, Dumollard JM, Peoc’h M. STAT6: a review of a signaling pathway implicated in various diseases with a special emphasis in its usefulness in pathology. Pathol Res Pract. (2021) 223:153477. doi: 10.1016/j.prp.2021.153477
136. Sharma M, Lu HY, Vaseghi-Shanjani M, Bel KLD, Fornes O, van der Lee R, et al. Human Germline Heterozygous Gain-of-Function STAT6 Variants Cause Severe Allergic Disease. medRxiv. (2022).
137. Suratannon N, Ittiwut C, Dik WA, Ittiwut R, Meesilpavikkai K, Israsena N, et al. A germline STAT6 gain-of-function variant is associated with early-onset allergies. J Allergy Clin Immunol. (2022) 151(2):565–71. doi: 10.1016/j.jaci.2022.09.028565-571.e9
138. Felgentreff K, Siepe M, Kotthoff S, von Kodolitsch Y, Schachtrup K, Notarangelo LD, et al. Severe eczema and hyper-IgE in loeys–dietz-syndrome—contribution to new findings of immune dysregulation in connective tissue disorders. Clin Immunol. (2014) 150:43–50. doi: 10.1016/j.clim.2013.11.008
139. Frischmeyer-Guerrerio PA, Guerrerio AL, Oswald G, Chichester K, Myers L, Halushka MK, et al. TGFβ receptor mutations impose a strong predisposition for human allergic disease. Sci Transl Med. (2013) 5:195ra94. doi: 10.1126/scitranslmed.3006448
140. Lyons JJ, Liu Y, Ma CA, Yu X, O’Connell MP, Lawrence MG, et al. ERBIN Deficiency links STAT3 and TGF-β pathway defects with atopy in humans. J Exp Med. (2017) 214:669–80. doi: 10.1084/jem.20161435
141. Schwerd T, Twigg SRF, Aschenbrenner D, Manrique S, Miller KA, Taylor IB, et al. A biallelic mutation in IL6ST encoding the GP130 co-receptor causes immunodeficiency and craniosynostosis. J Exp Med. (2017) 214:2547–62. doi: 10.1084/jem.20161810
142. Béziat V, Tavernier SJ, Chen YH, Ma CS, Materna M, Laurence A, et al. Dominant-negative mutations in human IL6ST underlie hyper-IgE syndrome. J Exp Med. (2020) 217:e20191804. doi: 10.1084/jem.20191804
143. Spencer S, Bal SK, Egner W, Allen HL, Raza SI, Ma CA, et al. Loss of the interleukin-6 receptor causes immunodeficiency, atopy, and abnormal inflammatory responses. J Exp Med. (2019) 216:1986–98. doi: 10.1084/jem.20190344
144. Barzaghi F, Passerini L. IPEX Syndrome: improved knowledge of immune pathogenesis empowers diagnosis. Front Pediatr. (2021) 9:612760. doi: 10.3389/fped.2021.612760
145. Zhang X, Olsen N, Zheng SG. The progress and prospect of regulatory T cells in autoimmune diseases. J Autoimmun. (2020) 111:102461. doi: 10.1016/j.jaut.2020.102461
146. Gambineri E, Ciullini Mannurita S, Hagin D, Vignoli M, Anover-Sombke S, DeBoer S, et al. Clinical, immunological, and molecular heterogeneity of 173 patients with the phenotype of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Front Immunol. (2018) 9:2411. doi: 10.3389/fimmu.2018.02411
147. Hull CM, Nickolay LE, Estorninho M, Richardson MW, Riley JL, Peakman M, et al. Generation of human islet-specific regulatory T cells by TCR gene transfer. J Autoimmun. (2017) 79:63–73. doi: 10.1016/j.jaut.2017.01.001
148. Ben-Skowronek I. IPEX Syndrome: genetics and treatment options. Genes (Basel). (2021) 12:323. doi: 10.3390/genes12030323
149. Maher MC, Hall EM, Horii KA. Generalized eczematous dermatitis and pruritus responsive to dupilumab in a patient with immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Pediatr Dermatol. (2021) 38:1370–1. doi: 10.1111/pde.14717
150. Schmetterer KG, Haiderer D, Leb-Reichl VM, Neunkirchner A, Jahn-Schmid B, Küng HJ, et al. Bet v 1–specific T-cell receptor/forkhead box protein 3 transgenic T cells suppress bet v 1–specific T-cell effector function in an activation-dependent manner. J Allergy Clin Immunol. (2011) 127:238–45. doi: 10.1016/j.jaci.2010.10.023
151. Passerini L, Bacchetta R. Forkhead-Box-P3 gene transfer in human CD4+ T conventional cells for the generation of stable and efficient regulatory T cells, suitable for immune modulatory therapy. Front Immunol. (2017) 8:1282. doi: 10.3389/fimmu.2017.01282
152. Moin A, Farhoudi AAH, Moein M, Pourpak Z, Bazargan N. Cutaneous manifestations of primary immunodeficiency diseases in children. Iran J Allergy Asthma Immunol. (2006) 5(3):121–6.
153. Berron-Ruiz A, Berron-Perez R, Ruiz-Maldonado R. Cutaneous markers of primary immunodeficiency diseases in children. Pediatr Dermatol. (2000) 17:91–6. doi: 10.1046/j.1525-1470.2000.01721.x
154. Castagnoli R, Lougaris V, Giardino G, Volpi S, Leonardi L, Torre FL, et al. Inborn errors of immunity with atopic phenotypes: a practical guide for allergists. World Allergy Organ J. (2021) 14:100513. doi: 10.1016/j.waojou.2021.100513
155. Guttman-Yassky E, Ungar B, Malik K, Dickstein D, Suprun M, Estrada YD, et al. Molecular signatures order the potency of topically applied anti-inflammatory drugs in patients with atopic dermatitis. J Allergy Clin Immunol. (2017) 140:1032–42. doi: 10.1016/j.jaci.2017.01.027
156. Bernardini R, Vespasiani GT, Giannetti A. An overview of off-label use of humanized monoclonal antibodies in paediatrics. Medicina (B Aires). (2022) 58:625. doi: 10.3390/medicina58050625
157. Guttman-Yassky E, Bissonnette R, Ungar B, Suárez-Fariñas M, Ardeleanu M, Esaki H, et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J Allergy Clin Immunol. (2019) 143:155–72. doi: 10.1016/j.jaci.2018.08.022
158. Paller AS, Beck LA, Blauvelt A, Siegfried EC, Cork MJ, Wollenberg A, et al. Infections in children and adolescents treated with dupilumab in pediatric clinical trials for atopic dermatitis—a pooled analysis of trial data. Pediatr Dermatol. (2022) 39:187–96. doi: 10.1111/pde.14909
159. Fujishima C, Munemoto S, Hioki C, Sasaki H, Yoshida H, Yamamoto T, et al. Successful dupilumab therapy for atopic dermatitis in a patient with X-linked agammaglobulinaemia. Eur J Dermatol. (2022) 32:416–7. doi: 10.1684/ejd.2022.4325
160. Nihal A, Comstock JR, Holland KE, Singh AM, Seroogy CM, Arkin LM. Clearance of atypical cutaneous manifestations of hyper-IgE syndrome with dupilumab. Pediatr Dermatol. (2022) 39(6):940–2. doi: 10.1111/pde.15072-40-942
161. Tanner JF, Knutsen AP, Siegfried E. Safety and efficacy of dupilumab in an atopic adolescent with primary immune deficiency. Dermatitis. (2021) 32:e91–92. doi: 10.1097/DER.0000000000000712
162. Patruno C, Potestio L, Scalvenzi M, Battista T, Raia F, Picone V, et al. Dupilumab for the treatment of adult atopic dermatitis in special populations. J Dermatol Treat. (2022) 33(7):3028–33. doi: 10.1080/09546634.2022.2102121
163. Chan S, Cornelius V, Cro S, Harper JI, Lack G. Treatment effect of omalizumab on severe pediatric atopic dermatitis: the ADAPT randomized clinical trial. JAMA Pediatr. (2020) 174:29–37. doi: 10.1001/jamapediatrics.2019.4476
164. Iannelli M, Caminiti L, Vaccaro M, Marafioti I, Spinuzza A, Panasiti I, et al. Omalizumab for treatment of refractory severe atopic dermatitis. A pediatric perspective. Dermatol Ther. (2020) 33:e13519. doi: 10.1111/dth.13519
165. Arasu A, Sharma N, Yazdabadi A, Sladden M. Dual biologic therapy with omalizumab and dupilumab for refractory atopic disease. Australas J Dermatol. (2022) 63:110–1. doi: 10.1111/ajd.13743
166. Barrios JL, Bégin P, Paradis L, Hatami A, Paradis J, Des Roches A. Anti-IgE therapy and severe atopic dermatitis: a pediatric perspective. J Am Acad Dermatol. (2013) 69:832–4. doi: 10.1016/j.jaad.2013.05.035
167. Furue M. Regulation of skin barrier function via competition between AHR axis versus IL-13/IL-4‒JAK‒STAT6/STAT3 axis: pathogenic and therapeutic implications in atopic dermatitis. J Clin Med. (2020) 9:3741. doi: 10.3390/jcm9113741
168. Boguniewicz M. Biologics for atopic dermatitis. Immunol Allergy Clin. (2020) 40:593–607. doi: 10.1016/j.iac.2020.06.004
169. FDA. Sanofi and Regeneron Announce FDA Approval of Dupixent® (dupilumab), the First Targeted Biologic Therapy for Adults with Moderate-to-Severe Atopic Dermatitis.
170. EMA. Dupixent® (dupilumab) approved by European Commission for Adolescents with moderate-to-severe atopic dermatitis.
171. FDA. FDA approves Dupixent® (dupilumab) as first biologic medicine for children aged 6 to 11 years with moderate-to-severe atopic dermatitis.
172. Keam SJ. Nemolizumab: first approval. Drugs. (2022) 82(10):1143–50. doi: 10.1007/s40265-022-01741-z
173. Japan’s MHLW. Japan’s MHLW Approves Pfizer’s CIBINQO® (abrocitinib) for Adults and Adolescents with Moderate to Severe Atopic Dermatitis.
174. Deeks ED, Duggan S. Abrocitinib: first approval. Drugs. (2021) 81(18):2149–57. doi: 10.1007/s40265-021-01638-3
175. Radi G, Simonetti O, Rizzetto G, Diotallevi F, Molinelli E, Offidani A. Baricitinib: the first jak inhibitor approved in Europe for the treatment of moderate to severe atopic dermatitis in adult patients. Healthcare. (2021):1575. doi: 10.3390/healthcare9111575
176. FDA. Incyte announces u. S. Fda approval of opzelura™(Ruxolitinib) cream, a topical jak inhibitor, for the treatment of atopic dermatitis(Ad) (2021) 9(11):1575.
Keywords: skin involvement, eczema, tailored treatment, biologics, inborn errors of immunity
Citation: Giancotta C, Colantoni N, Pacillo L, Santilli V, Amodio D, Manno EC, Cotugno N, Rotulo GA, Rivalta B, Finocchi A, Cancrini C, Diociaiuti A, El Hachem M and Zangari P (2023) Tailored treatments in inborn errors of immunity associated with atopy (IEIs-A) with skin involvement. Front. Pediatr. 11:1129249. doi: 10.3389/fped.2023.1129249
Received: 21 December 2022; Accepted: 3 March 2023;
Published: 22 March 2023.
Edited by:
Oded Shamriz, Hadassah Medical Center, IsraelReviewed by:
Marie-anne Morren, Centre Hospitalier Universitaire Vaudois (CHUV), SwitzerlandAntonio Condino-Neto, University of São Paulo, Brazil
Lovro Lamot, University of Zagreb, Croatia
© 2023 Giancotta, Colantoni, Pacillo, Santilli, Amodio, Manno, Cotugno, Rotulo, Rivalta, Finocchi, Cancrini, Diociaiuti, El Hachem and Zangari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola Zangari cGFvbGEuemFuZ2FyaUBvcGJnLm5ldA==
Specialty Section: This article was submitted to Pediatric Immunology, a section of the journal Frontiers in Pediatrics