 Xiao Li
Xiao Li Yu Tang1
Yu Tang1 Yuan Wang
Yuan Wang Weihua Zhang
Weihua Zhang Yuelin Shen
Yuelin Shen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 09 March 2023
Sec. Genetics of Common and Rare Diseases
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1118097
This article is part of the Research TopicRecent Advances in Understanding The Genetics of Immunological DisordersView all 12 articles
This report describes a case of a 22 months Chinese boy with COPA syndrome bearing the c.715G > C (p.A239P) genotype. In addition to interstitial lung diseae, he also suffered from recurrent chilblain-like rashes, which has not been previously reported, and neuromyelitis optica spectrum disorder (NMOSD), which is a very rare phenotype. Clinical manifestations expanded the phenotype of COPA syndrome. Notably, there is no definitive treatment for COPA syndrome. In this report, the patient has achieved short-term clinical improvement with sirolimus.
In 2019, the International Union of Immunological Societies Expert Committee (IUIS) classified COPA syndrome as a non-inflammasome-associated disease (1). COPA syndrome shares very similar symptoms and pathogenesis with another Type I interferonopathy, namely, STING-associated vasculopathy with onset in infancy (SAVI). STING and subsequent IFN pathway are activated by the mutation of STING1 gene. Up till now, a total of 78 individuals carrying 16 variants of COPA gene have been reported in the literature. COPA syndrome, also known as an immune deficiency disease with strong clinical heterogeneity and certain commonality (2), usually affects the lungs, kidneys, and joints. Skin vasculopathy is considered as a core feature of SAVI, which was observed in 77% of the reported patients, but has yet to be described in any patient with COPA syndrome. In this report, we present a pediatric case with recurrent chilblain-like rashes, which is a kind of skin vasculopathy. In addition, the patient also suffered from NMOSD, which has been reported only once before.
A 22-month-old boy presented with irritability for 8 days and limb weakness for 6 days, accompanied by lethargy and vomiting. The boy had a history of transient paralysis of the right upper limb after fever at the age of 19-month-old, and his symptoms alleviated three days later. Additionally, he also manifested repeated facial and ear rashes, which were diagnosed as chilblain. Physical examination showed decreased muscle strength (3+/5 and 3/5 for upper and lower limbs, respectively), mild hypertonia in the left lower limb, nuchal rigidity, normal deep reflex, negative Babinski's sign, Kernig's sign and Brudzinski's sign. Laboratory tests (Table 1) revealed positive perinuclear antineutrophil cytoplasmic antibody (pANCA) and antinuclear antibody (ANA) (1:320), negative anti-double-stranded DNA (ds-DNA) antibody and anti-Sm antibody, positive serum anti-aquaporin 4 antibody (AQP4) (1:1,000, cell based assay), negative serum anti-myelin oligodendrocyte glycoprotein antibody (MOG), anti-glial fibrillary acidic protein (GFAP) antibody and serum antibodies against autoimmune peripheral neuropathy (KingMed Diagnostics). No abnormalities was found in ophthalmic examination revealed. Routine and biochemical test of cerebrospinal fluid (CSF) were normal. CSF culture and metagenomic next-generation sequencing (mNGS) showed no etiology. Brain MRI showed lesions involving medulla and white matter around the ventricle and small encephalomalacia foci with gliosis in the left basal ganglia (Figure 1A). MRI of the optic nerve was normal (Figure 1B). MRI of the Spinal cord showed longitudinally extensive transverse myelitis lesions from the medulla to C6 level (Figures 1C,D). According to the NMOSD diagnostic criteria proposed by the NMO diagnostic Group in 2015 (2), the child was diagnosed as NMOSD (based on the existence of two core clinical symptoms, namely, acute myelitis and area postrema syndrome, and positive AQP4 antibody). The patient was reated with high dose of intravenous methylprednisolone (20 mg per kilogram for 4 day) followed by oral prednisone with gradual diminution of dose, along with intravenous immunogloblin (IVIG) (2 g/kg). Then his muscle strength gradually improved without recurrence. Disease-Modifying Treatment (DMT) is recommended as the standard treatment in remission of NMODS, which can reduce the clinical onset of patients with recurrent remission. Regrettably, his parents refused to use DMT in consideration of its adverse effects.
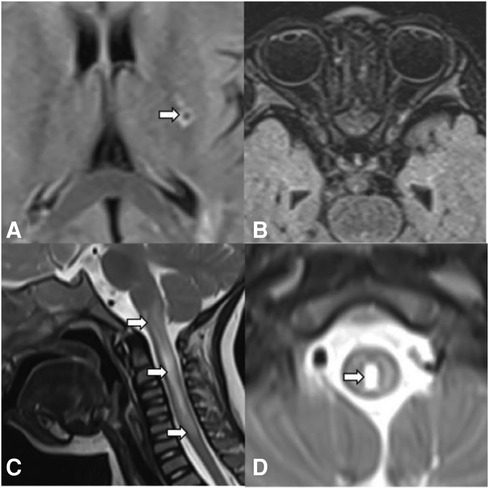
Figure 1. Brain MRI, spinal cord MRI, and optic MRI of the patient. Axial T2-weighted fluid-attenuated inversion recovery (FLAIR) MRI shows small encephalomalacia foci with gliosis in the left basal ganglia (A, arrows) and normal optic nerves (B). Sagittal (C) and axial (D) T2-weighted MRI of the spinal cord demonstrates longitudinally extensive transverse myelitis (LETM) lesion involving medulla to C6 leve (arrows).
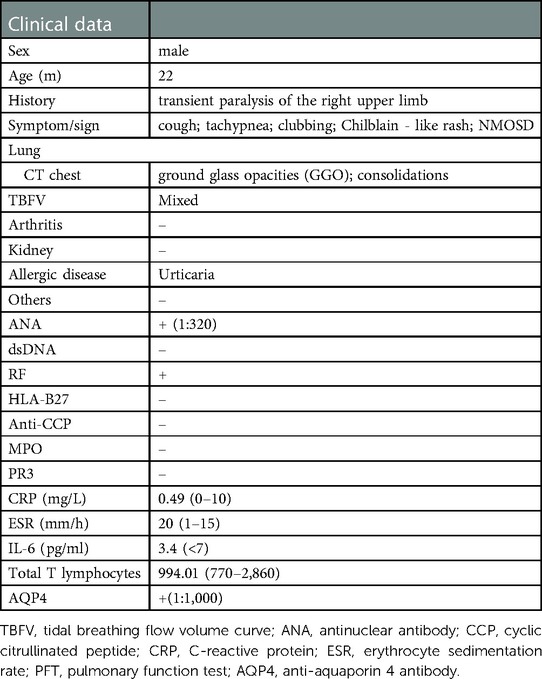
Table 1. Clinical data of the patient with COPA syndrome.
The patient developed dry cough, shortness of breath accompanied with rashes in the following months. Physical examination showed tachypnea, retractions and clubbing fingers, as well as Chilblain-like rashes on both of cheeks (Figure 2). His respiratory symptoms revealed a lousy response to anti-infection treatment and deteriorated. Chest CT (Figure 3) showed ground glass opacities (GGO) and consolidations in both lungs, suggesting interstitial lung disease (ILD). The whole exome sequencing (WES), conducted by Beijing MyGenostics Laboratory, revealed a heterozygous variant of COPA gene, that is, c.715G > C (p.A239P, NM_004371), which was confirmed by sanger sequencing (Figure 4). The patient was administrated with prednisone 2 mg/kg per day for one month at the age of 25 months. His tachypnea and cough were slightly alleviated but not disappear during the treatment. Then, he received a combined treatment of sirolimus with a dose of 0.8 mg/m2 at the age of 26 months. Meanwhile, the dosage of prednisone gradually decreased to 0.5 mg/kg and remained unchanged. Afterwards, his cough and tachypnea gradually disappeared. Repeated chest CT at the age of 29-months old showed significant absorption of GGO and consolidations in both lungs after the 4 months of prednisone treatment and 3 months of combined sirolimus treatment. However, the patient was non-adherent to follow up.
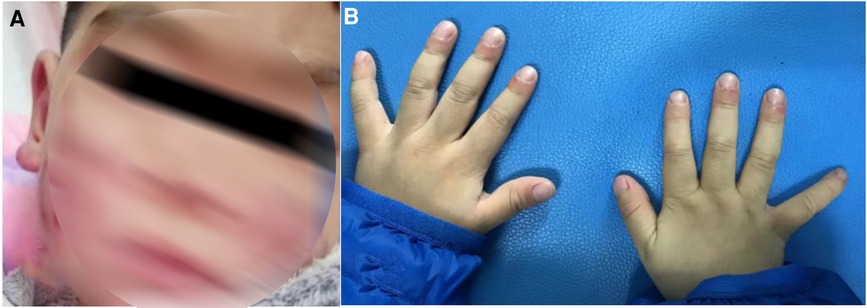
Figure 2. (A) Chilblain-like rash was observed on the face and ears of the child. (B) Clubbing fingers.

Figure 3. ①②: Chest CT before the treatment at the age of 25-months old revealing ground glass opacities (GGO) and consolidations in both lungs. ③④: Chest CT at the age of 29-months old revealing significant absorption of GGO and consolidations in the lung after treatment for 4 months.
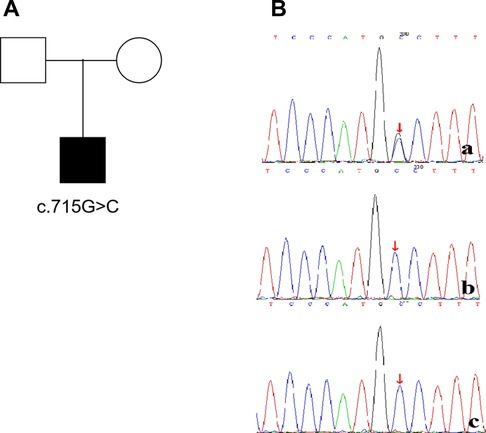
Figure 4. The child(a) has a de novo mutation (c.715G>C,p.A239P), his father(b) and mother(c) have no mutation at the same location.
COPA gene is located on chromosome1q23.2 and including 33 exons (54 kb). To date, most of the pathogenic COPA variants identified in patients with autoinflammatory diseases compatible with COPA syndrome are located in the 14 amino acid region and the WD40 domain with important functions, except for 4 mutations, that is c.433C > T (p.P145S) (3), c.596A > G (p.H199R) (4), c.841C > T (p.R281W) (5) and c.855G > C (p.Q285H) (6). The genetic testing of this patient revealed a variant of c.715G > C (p.A239P), located within the WD40 domain, which has been previously reported by Pamela Psarianos (7).
Clinically, COPA syndrome mainly manifests diffuse alveolar hemorrhage or interstitial lung disease, arthritis, and renal injury. Additionally, its onset is mostly in childhood, without racial predilection. Watkin et al. (8) reported that all of the patients had lung disease diagnosed as pulmonary hemorrhage, interstitial lung disease, or both. Arthritis was found in 95% of children, with the knee joint and interphalangeal joints being the most common, and rheumatoid factor positive in 43% of children. In the available literature, only 4 patients have neurological symptoms (9). Bader-meunier B et al. (6) reported a case of COPA syndrome with arthritics only. The mutation occurred on the outer surface of the adjacent blade within the seven-bladed b-propellor structure, distinctting from the previously reported hot spot mutations. To our great knowledge, there have been no previous reports of rashes in patients with COPA syndrome.
In this case, the child presented with limb weakness, and MRI of the spinal cord indicated patchy abnormal signal shadows from C1 to C6, which was consistent with acute myelitis. In terms of his clinical sympotom of vomiting, encephalitis was excluded due to no fever and convulsive seizure, normal CSF routine and biochemistry, no etiology of CSF culture and mNGS, negative antibodies against autoimmune peripheral neuropathy. Brain MRI suggested abnormal signal shadow in the medulla oblongata, area postrema syndrome was therefore taken into consideration. At the same time, according to the NMOSD diagnostic criteria formulated by the International NMO Diagnostic Group (IPND) in 2015, the child could be diagnosed as NMOSD due to positive AQP4. Additionally, this child was transiently paralyzed in his right upper limb at the age of 19 months. Combined with head MRI, the left basal ganglia encephalomalacia lesion was shown (there was no change in subsequent brain MRI), which might be episode of focal cerebral ischemia. As far as we known, NMOSD is a very rare phenotype of COPA syndrome. In the past, only one patient suffered from hearing loss due to bacterial meningitis and bilateral recurrent neuromyelitis optica. However, the presence of AQP4-Ab was not reported in this patient (10). NMOSD is an uncommon antibody-mediated central nervous system disease. Antibodies against aquaporin-4 (AQP4-Ab), a water channel expressed on astrocytes has been found in approximately 75% of patients. A recent whole-genome sequence study identified genetic variants in the major histocompatibility region that contribute to the etiology of NMOSD (11). About one quarter of AQP4-Ab positive NMOSD patients suffer from another autoimmune disease, such as systemic lupus erythematosus (SLE) and myasthenia gravis (12, 13). SLE and NMOSD share a common origin of interferonopathy. Intriguingly, Jac Williams (14) reported that either an increase in endogenous IFNα (such as in patients with SLE) or an increase in exogenous IFNα (treatment with recombinant interferon alpha) could promote the development of NMOSD. COPA syndrome is an autoinflammatory disease with autoimmune features, suggesting that the pathogenesis of COPA syndrome and NMOSD comorbidity may be similar to the SLE and NMOSD comorbidities, which may be associated with humoral immunity.
Patients with COPA syndrome shares similar clinical features with SAVI, such as ILD, joint and kidney involvement (15). Skin vasculopathy, ranging from a mild rash or livedo to severe ulcerative lesions and extensive tissue loss, is a core feature of SAVI, which is observed in 77% of the reported patients, but has yet to be described in any previous COPA syndrome patients. This is the first case of chilblain-like rashes in COPA syndrome.
At present, COPA syndrome treatment mainly refers to other autoimmune diseases. Common treatment schemes include glucocorticoids in combination with immunosuppressors such as JAK1/2 inhibitor (ruxolitinib, baricitinib). JAK inhibitors are widely used to inhibit cytokine signaling, including the downstream of interferons and other cytokines. It is reported that the application of JAK inhibitors can improve the patient's well-being and quality of life of the patients (16, 17). Glucocorticoid combined with hydroxychloroquine and mycophenolate mofetil (MMF) can also alleviate cough in COPA patients (7). In addition, Guan Y et al. (2) reported that two cases of COPA syndrome were treated with sirolimus and achieved favourable therapeutic effect. In the present report, the symptoms of the child have been remarkably alleviated after the treatment of glucocorticoid in combination with sirolimus, which supports the possibility that sirolimus may serve as an effective treatment for COPA syndrome. Blocking the mTOR pathway may be the mechanism of sirolimus in treating COPA syndrome, which is one of the downstream signaling pathways activated by STING. The long-term prognosis of COPA syndrome remains unclear due to the limited number of COPA syndrome cases and insufficient follow-up periods.
There are still several limitations in our study. Skin biopsy was not performed to verify that the chilblain-like rash in the child was vasculitis. Moreover, we only conducted short-term follow-up.
In summary, we first reported a new phenotype of skin involvement in one patient with COPA syndrome, which expanded the phenotypic spectrum of this disease.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Children's Hospital Affiliated to Zhengzhou University/Henan Children's Hospital/Zhengzhou Children's Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
XL and YW: supervised the patient care, conceptualized and designed the study, drafted the initial manuscript, and reviewed and revised the manuscript. YT and LZ: performed the lung scintigraphy, designed the study, and reviewed and revised the manuscript. YW and WZ: performed the nervous system scintigraphy, designed the study, and reviewed and revised the manuscript. YS: performed genetic analysis, designed the study, collected data, and reviewed and revised the manuscript. XT: conceptualized and designed the study, coordinated and supervised data collection, and critically reviewed the manuscript for important intellectual content. All authors contributed to the article and approved the submitted version.
National Natural Science Foundation of China, Youth Science Foundation Project (grant no. 82202061).
We are grateful to the patients and their family for their collaboration.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol. (2020) 40(1):24–64. doi: 10.1007/s10875-019-00737-x
2. Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J Exp Med. (2016) 213(12):2527–38. doi: 10.1084/jem.20161596
3. He T, Qi Z, Luo S, Xia Y, Li C, Huang Y, et al. Clinical and genetic analysis of COPA syndrome in one case. J Clin Pediatr. (2018) 36:787–90. doi: 10.3969/j.issn.1000-3606.2018.10.015
4. Guan Y, Liu H, Tang X, Xu H, Peng Y, Zhou C, et al. Effective sirolimus treatment of 2 COPA syndrome patients. J Allergy Clin Immunol Pract. (2021) 9(2):999–1001.e1. doi: 10.1016/j.jaip.2020.10.019
5. Zeng J, Hao J, Zhou W, Zhou Z, Miao H. A novel mutation c.841C>T in COPA syndrome of an 11-year-old boy: a case report and short literature review. Front Pediatr. (2021) 9:773112. doi: 10.3389/fped.2021.773112
6. Bader-Meunier B, Bustaffa M, Iskounen T, Carter E, Marsh JA, Baujat G, et al. Rheumatoid factor positive polyarticular juvenile idiopathic arthritis associated with a novel COPA mutation. Rheumatology. (2021) 60(5):e171–3. doi: 10.1093/rheumatology/keaa763
7. Psarianos P, Kwan JYY, Dell S, Wee WB, Rey-McIntyre K, Chen H, et al. COPA syndrome (Ala239Pro) presenting with isolated follicular bronchiolitis in early childhood: case report. J Clin Immunol. (2021) 41(7):1660–3. doi: 10.1007/s10875-021-01082-8
8. Watkin LB, Jessen B, Wiszniewski W, Vece TJ, Jan M, Sha Y, et al. COPA Mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat Genet. (2015) 47(6):654–60. doi: 10.1038/ng.3279
9. Frémond ML, Crow YJ. STING-mediated lung inflammation and beyond. J Clin Immunol. (2021) 41(3):501–14. doi: 10.1007/s10875-021-00974-z
10. Taveira-DaSilva AM, Markello TC, Kleiner DE, Jones AM, Groden C, Macnamara E, et al. Expanding the phenotype of COPA syndrome: a kindred with typical and atypical features. J Med Genet. (2019) 56(11):778–82. doi: 10.1136/jmedgenet-2018-105560
11. Estrada K, Whelan CW, Zhao F, Bronson P, Handsaker RE, Sun C, et al. A whole-genome sequence study identifies genetic risk factors for neuromyelitis optica. Nat Commun. (2018) 9(1):1929. doi: 10.1038/s41467-018-04332-3
12. Leite MI, Coutinho E, Lana-Peixoto M, Apostolos S, Waters P, Sato D, et al. Myasthenia gravis and neuromyelitis optica spectrum disorder: a multicenter study of 16 patients. Neurology. (2012) 78(20):1601–7. doi: 10.1212/WNL.0b013e31825644ff
13. Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation. (2012) 9:14. doi: 10.1186/1742-2094-9-14
14. Williams J, McGlasson S, Irani S, Duffy D, Crow Y, Hunt D, et al. Scottish NMOSD study group, et al. Neuromyelitis optica in patients with increased interferon alpha concentrations. Lancet Neurol. (2020) 19(1):31–3. doi: 10.1016/S1474-4422(19)30445-4
15. Volpi S, Tsui J, Mariani M, Pastorino C, Caorsi R, Sacco O, et al. Type I interferon pathway activation in COPA syndrome. Clin Immunol. (2018) 187:33–6. doi: 10.1016/j.clim.2017.10.001
16. Frémond M-L, Legendre M, Fayon M, Clement A, Filhol-Blin E, Richard N, et al. Use of ruxolitinib in COPA syndrome manifesting as life-threatening alveolar haemorrhage. Thorax. (2020) 75(1):92–5. doi: 10.1136/thoraxjnl-2019-213892
Keywords: children, copa syndrome, neuromyelitis optica spectrum disorder, rashes, sirolimus
Citation: Li X, Tang Y, Zhang L, Wang Y, Zhang W, Wang Y, Shen Y and Tang X (2023) Case report: COPA syndrome with interstitial lung disease, skin involvement, and neuromyelitis spectrum disorder. Front. Pediatr. 11:1118097. doi: 10.3389/fped.2023.1118097
Received: 7 December 2022; Accepted: 20 February 2023;
Published: 9 March 2023.
Edited by:
Jinqiao Sun, Fudan University, ChinaReviewed by:
Tiphanie Phillips Vogel, Baylor College of Medicine, United States© 2023 Li, Tang, Zhang, Wang, Zhang, Wang, Shen and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaolei Tang dGFuZ3hsOUBnbWFpbC5jb20=
Specialty Section: This article was submitted to Genetics of Common and Rare Diseases, a section of the journal Frontiers in Pediatrics
Abbreviations NMOSD: neuromyelitis optica spectrum disorder; AQP4, anti-aquaporin 4 antibody; IFN, interferon; MRI, magnetic resonance imaging; pANCA, perinuclear antineutrophil cytoplasmic antibody; ANA, antinuclear antibody; MOG, anti-myelin oligodendrocyte glycoprotein antibody; GFAP, anti-glial fibrillary acidic protein; ILD, interstitial lung disease; SLE, systemic lupus erythematosus; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.