 Fen Lu
Fen Lu Xin Xu
Xin Xu Bixia Zheng2
Bixia Zheng2 Chunli Wang
Chunli Wang Wei Zhou
Wei Zhou- 1Department of Rehabilitation, Children's Hospital of Nanjing Medical University, Nanjing, China
- 2Nanjing Key Laboratory of Pediatrics, Children's Hospital of Nanjing Medical University, Nanjing, China
Biallelic TENM3 variants were recently reported to cause non-syndromic microphthalmia with coloboma-9 (MCOPCB9) and microphthalmia and/or coloboma with developmental delay (MCOPS15). To date, only eight syndromic and non-syndromic microphthalmia cases with recessive TENM3 variants have been reported. Herein, we report two unrelated new cases with biallelic variants in TENM3, widening the molecular and clinical spectrum. Regarding patient 1, WES revealed compound heterozygous variants in the TENM3 gene: c.3847_3855del; p.Leu1283_Ser1285del and c.3698_3699insA; p.Thr1233Thrfs*20 in the index patient, who was presenting with bilateral microphthalmia, congenital cataract, microcephaly, and global developmental delay. Regarding patient 2, compound missense heterozygous variants in the TENM3 gene were identified: c.941C > T; p.Ala314Val and c.6464T > C; p.Leu2155Pro in the 3-year-old boy, who presented with congenital esotropia, speech delay, and motor developmental delay. The clinical features of these two cases revealed high concordance with the previously reported cases, including microphthalmia and developmental delay. The presence of microcephaly in our patient potentially expands the neurologic phenotype associated with loss of function variants in TENM3, as microcephaly has not previously been described. Furthermore, we present evidence that missense variants in TENM3 are associated with similar, but milder, ocular features.
Introduction
Teneurin transmembrane protein 3 (TENM3) encodes a large transmembrane protein involved in neural development by regulating the establishment of proper connectivity within the nervous system (1–3). It has been found to play a role in the development of the human eye by regulating the formation of ipsilateral retinal mapping to both the dorsal lateral geniculate nucleus and the superior colliculus (4–6). The homozygous null variant was first reported in a Saudi Arabian consanguineous family with non-syndromic bilateral colobomatous microphthalmia (7). Subsequently, very few publications have reported patients with TENM3 variant-related syndromic microphthalmia to date (8–10). Here, we present two patients with recessive variants in TENM3, and we describe their clinical presentations, providing further clinical and molecular delineation of the TENM3 syndrome.
Materials and methods
Study participants
Following informed consent, we obtained pedigree information, clinical data, and blood samples from the families. We obtained approval for human subject research from the ethics committee of the Children's Hospital of Nanjing Medical University.
Whole exome sequencing
Trio-based WES was performed as previously described (11). In brief, genomic DNA was isolated from blood lymphocytes using the DNA isolation kit (Tiangen, China). Genomic DNA was sheared into fragments and then hybridized with the xGen Exome Research Panel v1.0 probe sequence capture array from IDT (Integrated Device Technology, United States) to enrich the exonic region. The enriched libraries were analyzed on an Illumina HiSeq XTen (Illumina, United States) platform. Low-quality variations of the quality score <20 (Q20) were filtered out. Sequencing reads were mapped to the GRCh37/Hg19 reference genome via Burrows-Wheeler Aligner (BWA) software. Single nucleotide variation (SNV) and inserts and deletions (INDEL) were filtered using GATK software (https://software.broadinstitute.org/gatk/). All identified variants were filtered using the 1000 Genomes Project (Chinese), dbSNP, Genome Aggregation Database (gnomAD), and ExAC database. Variants with a minor allele frequency higher than 5% were filtered out. Finally, the candidate variants were evaluated using the ACMG (American College of Medical Genetics and Genomics) criteria and further validated by direct Sanger sequencing.
TA cloning of mutant PCR products
The two heterozygous TENM3 variants in family 1 were both located in exon 19. To obtain a clean Sanger sequence of the two heterozygous variants, we cloned 383 bp-long PCR products of TENM3 exon19 using the pCR2.1-TOPO plasmid vector system (Invitrogen). PCR products were generated using TENM3 forward primer 5-ATCCTCAGCGTCAGGCAAGGAA-3 and reverse primer 5-TCCCCTGTCCCTGCGACGAC-3. The TA clone sequencing was conducted as previously described (12).
Consideration of structural data and evolutionary conservation for variant evaluation
Protein domain structure depictions and evaluation were based on the UniProt (Universal Protein Resource) database. Orthologous proteins used to evaluate evolutionary conservation were obtained from the Ensemble Genome Browser and were aligned using the Clustal Omega multiple sequence alignment tool (EMBL-EBI). And we evaluated the crystal structure of the two missense TENM3 variants in patient 2 using the online server, UCSF ChimeraX (http://www.cgl.ucsf.edu/chimerax//).
Results
Clinical findings
Patient 1 was the 5-month-old daughter of Chinese non-consanguineous parents. She had two older brothers and both are healthy. She was born at 39 weeks of gestation. Her birth weight was 2.7 kg (−1.46 SD) and her length was 40 cm (−5.89 SD). At birth, she was diagnosed with bilateral microphthalmia and congenital cataract and she appeared to have pendular nystagmus and esotropia (Figure 1B). She first visited our department of rehabilitation at the age of 5 months for developmental delay. Her developmental milestones were delayed. Her head control was unstable and she was unable to turn over and grab the toy on her chest. On careful physical examination, her height, weight, and head circumference were 57 cm (−4.03 SD), 5 kg (−3.75 SD), and 37 cm (−4.28 SD), respectively. Her prominent and low-set ears were noted (Figure 1B). Fundus examination revealed the posterior pole of the retina colobomas involving the optic discs and the fovea (Figure 1C). Her hearing assessment was normal. According to the Gesell Developmental Diagnostic Scale for children, the proband's gross motor skills indicated a developmental age of 8 weeks; the fine motor skills indicated a developmental age of 8 weeks; Her blood counts, liver and renal function tests, thyroid profile, and metabolic screen by mass spectrometry were normal. Brain magnetic resonance imaging (MRI) showed no structure malformations except a widening in the frontotemporal extracerebral space. Her mother had left exotropia and graduated from middle school with poor grades.

Figure 1. Trio-based WES identified compound heterozygous variants (p.Leu1283_Ser1285del; p.Thr1233Thrfs*20) in TENM3 in a patient with bilateral microphthalmia, global developmental delay, and microcephaly. (A) Pedigree and genotype information for members of family 1. Squares indicate males, circles indicate females, filled symbols indicate affected individuals, and open symbols indicate healthy individuals. Patient 1 is denoted by a black arrow. The proband carried compound heterozygous variants: c.3847_3855del; p.Leu1283_Ser1285del and c.3698_3699insA; p.Thr1233Thrfs*20. The mother (II-2) with heterozygosity of the p.Thr1233Thrfs*20 variant had left exophthalmia and graduated from middle school with poor grades. (B) Facial picture of the proband at the age of 5 months with microphthalmia, prominent and low-set ears, and microcephaly. (C) Fundus examination of patient 1 revealed the posterior pole of the retina colobomas involving the optic discs and the fovea. (D) A TA clone sequencing from the genomic DNA of patient 1 including the fragment of exon 19 showed the two variants: c.3847_3855del; p.Leu1283_Ser1285del and c.3698_3699insA; p.Thr1233Thrfs*20. WT, wild type; MUT, mutant type.
Patient 2 was the 3-year-and-5-month-old son of Chinese non-consanguineous parents. His weight was 17 kg (0.8 SD) and his height was 101 cm (0.2 SD). At birth, it was noted that he had bilateral esotropia. At the age of 3 years old, his bilateral esotropia was resolved (Figure 2B). What's more, his anterior segment and fundus examination showed no structural anomalies (Figure 2C). He had astigmatism in both eyes (−1.25 DC). He had mild motor delay. He was able to walk without support at 17 months. He had significant speech delay, not producing any meaningful words at 2 years and 5 months and speaking a few simple words (about 10 words) at 3 years and 5 months. He showed poor eye contact and was not interested in his surroundings, so his social interaction was abnormal. According to the Gesell Developmental Diagnostic Scale for children, the proband's delayed speech indicated a developmental age of only 18 months. He was also evaluated by the Autistic Behavior Checklist (ABC) and Childhood Autism Rating Scale (CARS) (ABC: score 40; CARS: score 32). His blood counts, liver and renal function tests, thyroid profile, and metabolic screen by mass spectrometry were normal. His hearing evaluation was also normal. His prominent and big ears were noted. A brain MRI did not show any intracranial abnormalities. His father had no abnormal eye appearance, but his eyes were myopic (−7.00 DS), and his mother was healthy. His grandfather had a history of fundus abnormality.
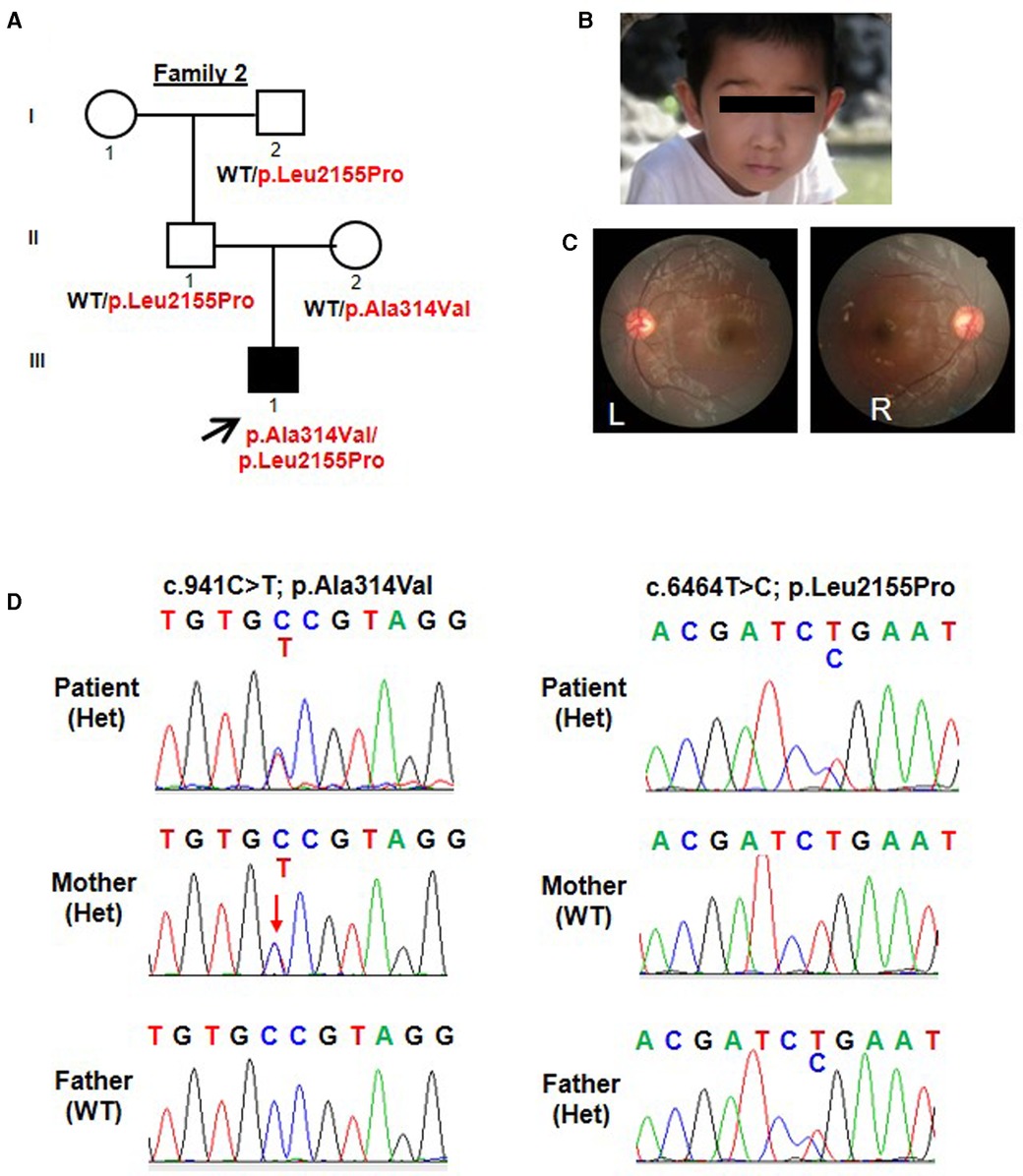
Figure 2. Trio-based WES identified compound heterozygous variants (p.Ala314Val; p.Leu2155Pro) in TENM3 in a patient with speech delay and motor developmental delay. (A) Pedigree and genotype information on members of family 2. Squares indicate males, circles indicate females, filled symbols indicate affected individuals, and open symbols indicate healthy individuals. Patient 2 is denoted by a black arrow. The proband carried compound heterozygous variants: c.941C > T; p.Ala314Val and c.6464T > C; p.Leu2155Pro. (B) Facial picture of the proband at the age of 3 years old with resolved esotropia. His prominent and big ears were noted. (C) Fundus examination of patient 2 showed no structural anomalies. (D) Sequencing chromatograms of the compound heterozygous TENM3 variants (c.941C > T; p.Ala314Val and c.6464T > C; p.Leu2155Pro) in patient 2 and the parents. WT, wild type; Het, heterozygous.
Genetic analysis
Initial genetic testing for patient 1 was carried out via WES, revealing two heterozygous variants in the TENM3 gene (Genbank association number: NM_001080477): c.3847_3855del; p.Leu1283_Ser1285del and c.3698_3699insA; p.Thr1233Thrfs*20 (Figure 1A). Sanger sequencing of the parents confirmed that the variant p.Leu1283_Ser1285del was inherited maternally. The variant p.Thr1233Thrfs*20 was confirmed to be de novo. The two variants were both located in exon 19. A TA clone sequencing including the fragment of exon 19 demonstrated that the two variants occurred biallelically (Figure 1D). The two variants were absent from the control database gnomAD. Based on the American College of Medical Genetics and Genomics (ACMG) guidelines, the variant c.3698_3699insA; p.Thr1233Thrfs*20 can be categorized as pathogenic (PVS1 + PM2 + PP4) and the variant c.3847_3855del; p.Leu1283_Ser1285del can be categorized as a variant of likely pathogenic (PM2 + PM3 + PM4).
Similarly, patient 2 had a WES that revealed compound heterozygous variants of the TENM3 gene, c.941C > T; p.Ala314Val and c.6464T > C; p.Leu2155Pro (Figure 2A). Sanger sequencing confirmed that his mother carried the c.941C > T (p.Ala314Val) mutation and his father carried the c.6464T > C (p.Leu2155Pro) mutation (Figure 2D). The p.Ala314Val variant was absent from the gnomAD. The p.Leu2155Pro variant occurred once heterozygously in the gnomAD. Both the missense changes yielded predominantly deleterious prediction scores using five algorithms (Polyphen2_HDIV, MutationTaster, SIFT, Provean, and REVEL); the predicted results can be found in the Supplementary Material.
The Ala314 change was located in the transmembrane domain and was evolutionarily well-conserved from Homo sapiens to zebrafish (Figures 3A,B). The Leu2155 residue was located in the YD-repeats domain and was well-conserved to zebrafish as well (Figures 3A,B). According to the ACMG guidelines, both variants, c.941C > T; p.Ala314Val and c.6464T > C; p.Leu2155Pro, can be categorized as variants of unknown significance (PM2 + PP2 + PP3). In evaluating the deleteriousness of the two missense variants in patient 2, three-dimensional structural modeling of the TENM3 protein showed that the mutations did not change the hydrogen bonding in the protein (blue), but repulsive force (purple) was generated between the R group of amino acid side chain and other nearby groups, which is unfavorable to the folding of the active protein and results in protein conformational instability (Figure 4).
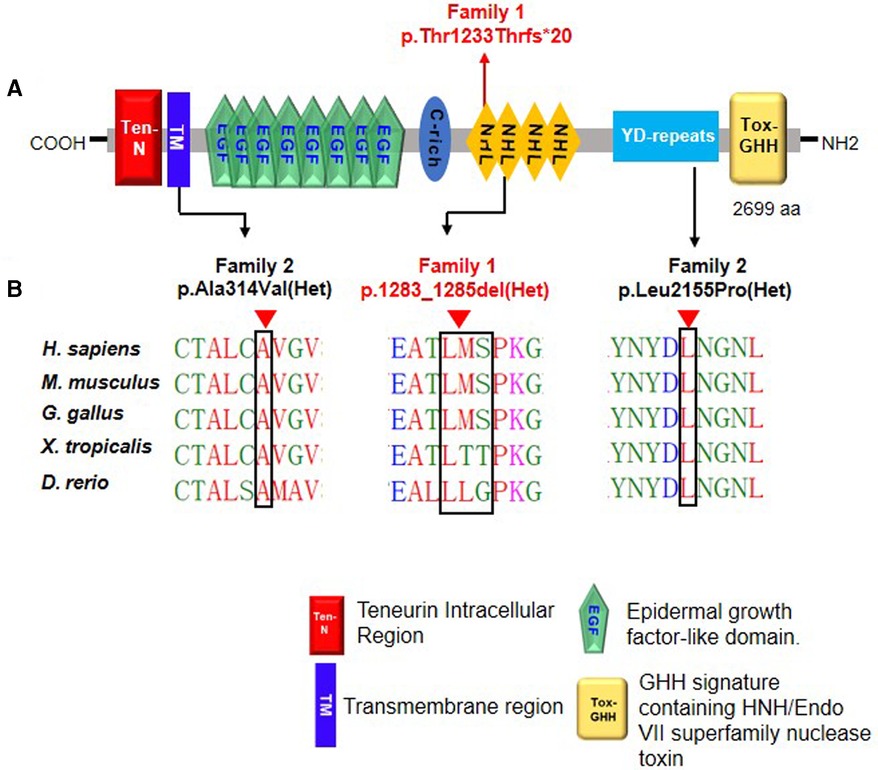
Figure 3. Schematic diagram of TENM3 functional domains and TENM3 variants identified in this study. (A) Depicts the protein domain structure of human TENM3 showing the domain position of the index heterozygous TENM3 variants. aa, amino acids. (B) Evolutionary conservation of amino acid position Ala1283-Ser1285 and Leu2155 in TENM3 protein across evolution.
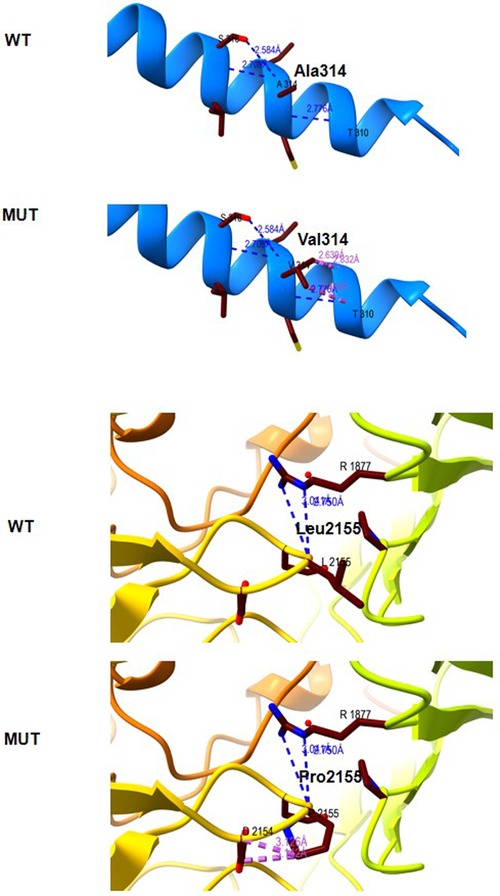
Figure 4. Molecular modeling of the wild type (WT) and mutant TENM3 protein (Mut). Three-dimensional structural modeling of the TENM3 protein showed that the mutations did not change the hydrogen bonding in protein (blue), but repulsive force (purple) was generated between the R group of amino acid side chain and other nearby groups.
Discussion
To date, only eight patients with recessive TENM3 variants have been described; information on the reported patients is shown in Table 1. The non-syndromic microphthalmia cases with recessive TENM3 variants only presented with moderate or severe eye abnormalities, including microphthalmia, microcornea, and retinal and iris coloboma, while TENM3 syndromic cases had additional abnormalities, such as craniofacial, renal, genital, cardiac, brain, and skeletal (9, 13, 14). To expand the TENM3 gene-related phenotypic spectrum, we describe the clinical features of two Chinese patients with compound heterozygous variants in TENM3. The main characteristic feature of this syndrome is eye involvement (15). All previously reported patients presented with moderate or severe eye abnormalities, including colobomatous microphthalmia, ocular coloboma, and cataract (9, 13, 14). The ocular features of patient 1 in our study are highly consistent with previous reports. However, patient 2, who harbored two missense variants (c.941C > T; p.Ala314Val and c.6464T > C; p.Leu2155Pro), did not have a phenotype related to microphthalmia, microcornea, or iris coloboma. At birth, it was noted that he had bilateral esotropia. However, his bilateral esotropia was resolved at the age of 3 years old. Furthermore, his anterior segment and fundus examinations showed normal structure. A literature review revealed that all variants in previously reported cases with microphthalmia and/or coloboma with developmental delay had biallelic truncating variants. The only reported case with compound heterozygote missense likely pathogenic sequence variations in TENM3 (p.Ala1349Gly and p.Arg2563Trp) showed right eye microphthalmia, sclerocornea of both eyes, anterior segment dysgenesis, and intellectual disability (14). We speculate that the missense mutations in patient 2 may have a mild effect on the structure of the TENM3 protein, which is associated with mild ocular symptoms. However, this must be further verified by in vitro experiments or animal experiments.
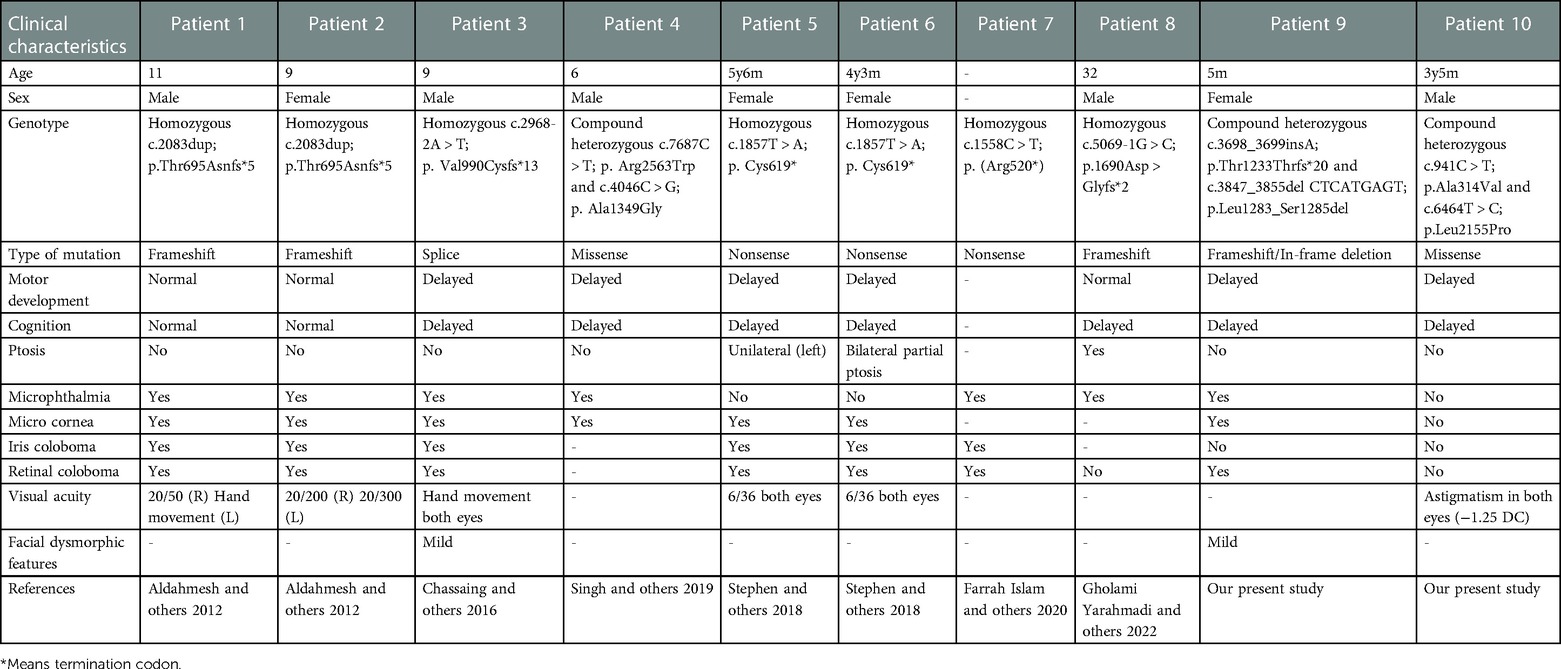
Table 1. Clinical manifestations of patients with variants in TENM3.
The mother of patient 1 with heterozygosity of the p.Thr1233Thrfs*20 variant had left exotropia and graduated from middle school with poor grades. The gnomAD constraint metric of TENM3 for loss of function is 1.0, indicating a high intolerance for heterozygous loss of function variants. However, no neurologic or ocular phenotypes were reported in individuals harboring a heterozygous allele in TENM3.
The two patients in our study had delayed developmental milestones similar to those observed in patients with recessive TENM3 variants: these included global developmental delay, speech delay, and motor developmental delay (8). Brain MRIs showed no structural abnormalities. Notably, the presence of microcephaly in patient 1 potentially expands the neurologic phenotype associated with loss of function variants in TENM3, as microcephaly has not previously been described. The two patients received rehabilitation training in our department and their motor function and language skills both improved, but there was no improvement in their eye symptoms.
In conclusion, we reported the clinical features of two cases with recessive variants in TENM3. While the majority of TENM3 syndromic or non-syndromic cases are truncating, missense variants have been described much less. It should be noted that biallelic missense variants in TENM3 seem to have a minor impact on eye involvement.
Data availability statement
The data presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by the ethics committee of Children's Hospital of Nanjing Medical University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
FL and XX collected the clinical data of the patients. FL, JT, and XZ conceived the project. BZ, CW, and WZ analyzed the result. JT and XZ wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
Acknowledgments
We would like to thank the families and study participants for their contributions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2023.1111771/full#supplementary-material.
References
1. Ben-Zur T, Feige E, Motro B, Wides R. The mammalian odz gene family: homologs of a drosophila pair-rule gene with expression implying distinct yet overlapping developmental roles. Dev Biol. (2000) 217(1):107–20. doi: 10.1006/dbio.1999.9532
2. Berns DS, DeNardo LA, Pederick DT, Luo L. Teneurin-3 controls topographic circuit assembly in the hippocampus. Nature. (2018) 554(7692):328–33. doi: 10.1038/nature25463
3. Takano I, Takeshita N, Yoshida M, Seki D, Oyanagi T, Kimura S, et al. Ten-m/Odz3 regulates migration and differentiation of chondrogenic ATDC5 cells via RhoA-mediated actin reorganization. J Cell Physiol. (2021) 236(4):2906–19. doi: 10.1002/jcp.30058
4. Carr OP, Glendining KA, Leamey CA, Marotte LR. Retinal overexpression of ten-m3 alters ipsilateral retinogeniculate projections in the wallaby (Macropus eugenii). Neurosci Lett. (2014) 566:167–71. doi: 10.1016/j.neulet.2014.02.048
5. Glendining KA, Liu SC, Nguyen M, Dharmaratne N, Nagarajah R, Iglesias MA, et al. Downstream mediators of ten-m3 signalling in the developing visual pathway. BMC Neurosci. (2017) 18(1):78. doi: 10.1186/s12868-017-0397-5
6. Young TR, Bourke M, Zhou X, Oohashi T, Sawatari A, Fassler R, et al. Ten-m2 is required for the generation of binocular visual circuits. J Neurosci. (2013) 33(30):12490–509. doi: 10.1523/JNEUROSCI.4708-12.2013
7. Aldahmesh MA, Mohammed JY, Al-Hazzaa S, Alkuraya FS. Homozygous null mutation in ODZ3 causes microphthalmia in humans. Genet Med. (2012) 14(11):900–4. doi: 10.1038/gim.2012.718.
8. Chassaing N, Ragge N, Plaisancie J, Patat O, Genevieve D, Rivier F, et al. Confirmation of TENM3 involvement in autosomal recessive colobomatous microphthalmia. Am J Med Genet A. (2016) 170(7):1895–8. doi: 10.1002/ajmg.a.37667
9. Stephen J, Nampoothiri S, Kuppa S, Yesodharan D, Radhakrishnan N, Gahl WA, et al. Novel truncating mutation in TENM3 in siblings with motor developmental delay, ocular coloboma, oval cornea, without microphthalmia. Am J Med Genet A. (2018) 176(12):2930–3. doi: 10.1002/ajmg.a.40658
10. Islam F, Htun S, Lai L-W, Krall M, Poranki M, Martin P-M, et al. Exome sequencing in patients with microphthalmia, anophthalmia, and coloboma (MAC) from a consanguineous population. Clin Genet. (2020) 98(5):499–506. doi: 10.1111/cge.13830
11. Wang C, Han Y, Zhou J, Zheng B, Zhou W, Bao H, et al. Splicing characterization of CLCNKB variants in four patients with type III bartter syndrome. Front Genet. (2020) 11:81. doi: 10.3389/fgene.2020.00081
12. Wang C, Chen Y, Zheng B, Zhu M, Fan J, Wang J, et al. Novel compound heterozygous CLCNKB gene mutations (c.1755A>G/c.848_850delTCT) cause classic bartter syndrome. Am J Physiol Renal Physiol. (2018) 315(4):F844–51. doi: 10.1152/ajprenal.00077.2017
13. Yarahmadi SG, Sarlaki F, Morovvati S. Novel mutation in TENM3 gene in an Iranian patient with colobomatous microphthalmia. Clin Case Rep. (2022) 10(3):e05532. doi: 10.1002/ccr3.5532
14. Singh B, Srivastava P, Phadke SR. Sequence variations in TENM3 gene causing eye anomalies with intellectual disability: expanding the phenotypic spectrum. Eur J Med Genet. (2019) 62(1):61–4. doi: 10.1016/j.ejmg.2018.05.004
Keywords: syndromic microphthalmia, TENM3, whole exome sequencing, genotype-phenotype, children
Citation: Lu F, Xu X, Zheng B, Wang C, Zhou W, Tang J and Zhao X (2023) Case report: Expansion of phenotypic and genotypic data in TENM3-related syndrome: Report of two cases. Front. Pediatr. 11:1111771. doi: 10.3389/fped.2023.1111771
Received: 30 November 2022; Accepted: 6 February 2023;
Published: 24 February 2023.
Edited by:
Xiu-An Yang, Chengde Medical College, ChinaReviewed by:
Emilia Severin, Carol Davila University of Medicine and Pharmacy, RomaniaMagdalena Budisteanu, Prof. Dr. Alexandru Obregia Psychiatry Hospital, Romania
© 2023 Lu, Xu, Zheng, Wang, Zhou, Tang and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoke Zhao eGlhb2tlemhhb0Buam11LmVkdS5jbg== Jian Tang dGFuZ2ppYW4yMDIwMDA4QDEyNi5jb20=
†These authors have contributed equally to this work
Specialty Section: This article was submitted to Genetics of Common and Rare Diseases, a section of the journal Frontiers in Pediatrics