 Caiyun Luo
Caiyun Luo Liucheng Yang
Liucheng Yang Kai Wu
Kai Wu- Department of Pediatric Surgery, Zhujiang Hospital, Southern Medical University, Guangzhou, China
Objective: This article aims to explore the diagnosis, molecular characteristics, treatment, and prognosis of epidermolysis bullosa with pyloric atresia (EB-PA).
Methods: The clinical manifestations, diagnosis and treatment, and genetic characteristics of a patient with EB-PA admitted to our hospital were analysed. The disease subtypes, concomitant abnormalities, molecular characteristics, and prognosis of patients with EB-PA were summarized by searching the EB-PA-related literature since 2011.
Results: We present a very low birth weight female infant with skin blisters and pyloric obstruction. Exome sequencing revealed heterozygous mutations in the ITGB4 gene: c.794dupC (p. S265fs*5) and c.2962G > A (p.A988T). This infant was diagnosed with EB-PA. Coverage of the wounds and Penicillin were used to prevent infection, but the patient eventually developed severe sepsis. A literature review was carried out including 49 cases of EB-PA; among these cases, 34 were preterm infants, weighing between 930 and 3,640 g. Of these EB-PA patients, 28 had accompanying malformations, including urinary system malformations and aplasia cutis congenita (ACC). Thirty-two patients identified the subtype of EB-PA, of whom 25 were diagnosed with junctional epidermolysis bullosa (JEB), 6 with epidermolysis bullosa simplex (EBS), and 1 with dystrophic epidermolysis bullosa (DEB). Genetic testing was conducted on 23 patients, of whom 15 carried Integrin Beta-4 (ITGB4) gene mutations and one JEB patient carried an Integrin Alpha-6 (ITGA6) gene mutation; 4 of the 5 EBS patients had Plectin (PLEC) gene mutations, and the other had an ITGB4 mutation. ITGB4 mutation cases involved 29 mutation sites, primarily concentrated in the region encoding the integrin beta subunit; PLEC mutation cases involved 7 mutation sites. Among all cases, 43 underwent pyloric atresia surgery, of whom 24 died postoperatively, and 6 without surgery therapy died within a short period.
Conclusion: EB-PA is a rare genetic disorder characterized by increased skin fragility and PA involving mutations in the ITGB4, PLEC, or ITGA6 genes. EB-PA has a high incidence of complications and mortality, surgery and supportive therapy are currently the most common treatment options.
1. Introduction
Epidermolysis bullosa (EB) is a group of inherited bullous diseases with structural abnormalities caused by genetic mutations of various structural proteins in the skin. Due to the increased fragility of the skin mucosa, mild trauma can cause tension bullous and skin erosion. Based on data collected by the U.S. National Epidermolysis Blister Registry from 1986 to 2002, the incidence of EB is approximately 20 per 1 million live births, and the prevalence is approximately 11 per 1 million live births (1). According to the latest International Consensus Meeting on EB, EB is divided into four types: EB simplex (EBS), junctional EB (JEB), dystrophic EB (DEB) and Kindler EB (KEB) (2). EB leading experts proposed an “onion skin” approach to classification, which further subclassified the four main types of EB into other subtypes by considering their phenotype (severity and distribution), mode of transmission, ultrastructural site of cleavage and associated findings, proteins involved (with or without specific immunofluorescence mapping findings listed), genes involved and mutational type, and specific mutation present (3).
Epidermolysis bullosa with pyloric atresia (EB-PA) is a rare clinical subtype of epidermolysis bullosa (EB) accompanied by high mortality. The diagnosis and management of EB-PA are difficult (4). In this study, we reviewed the clinical data of an infant admitted to our hospital and comprehensively analysed the characteristics of the cases in the literature to summarize the experience in diagnosing and treating EB-PA.
2. Case report
A female preterm infant, the first child of healthy nonconsanguineous parents, was born at 30 + 2 weeks of gestation by normal delivery, with a birth weight of 1,210 g and a negative family history. After birth, multiple skin defects were found in the whole body, mainly in the lower limbs. Physical examination at birth indicated skin defects of both lower limbs with basal flushing and exudation, but no obvious signs of infection were found in the wounds (Figure 1). The dorsum of the foot was a hyperflexion deformity with low muscle tone in the extremities. The upper abdomen was distended with a distinct gastric pattern. Routine blood test was measured and revealed that white blood cell count (WBC) 10.53 × 109/L (reference range 15.0–20.0 × 109/L), neutrophil numerals (Neut#) 8.13 × 109/L (ref. 1.8–6.3 × 109/L), haemoglobin (Hb) 137 g/L (ref. 170–200 g/L), platelets (PLT) 230 × 109/L (ref. 125–350 × 109/L). The results of coagulation function showed prothrombin time (PT) 16.0 s (ref. 9.8–12.1 s), active partial thromboplastin time (APTT) 110.2 s (ref. 25.0–31.3 s), international normalized ratio (INR) 1.38 (ref. 0.88–1.08). x-ray showed gas in the stomach but no inflation in the small intestine, which is considered pyloric obstruction (Figure 2A). Contrast medium accumulation in the stomach lumen was observed on upper gastrointestinal angiography, with no development in the duodenum or distal part (Figure 2B). There were no abnormalities in the respiratory, cardiac or urinary systems. A skin biopsy was recommended to further confirm the diagnosis of EB, but her parents refused to perform this test.
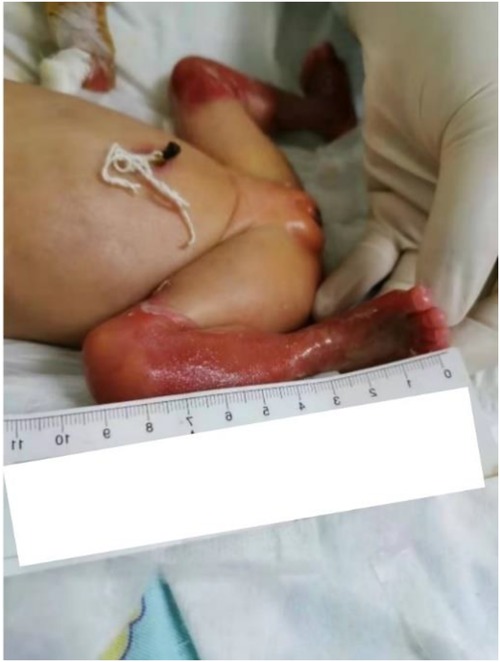
Figure 1. Severe ruptured blisters with skin lesions on both lower limbs.
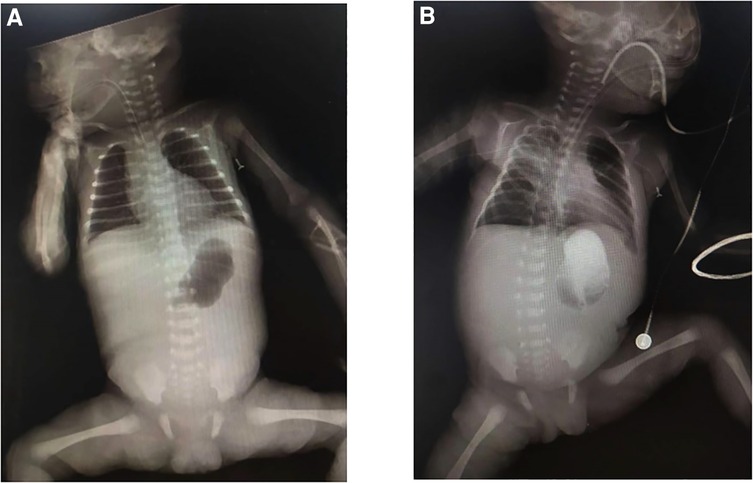
Figure 2. (A) x-ray demonstrates a single bubble sign; (B) upper gastrointestinal angiography shows no contrast uptake beyond the stomach.
The patient's clinical manifestations and examinations indicated a genetic disease. After obtaining consent from the patient's family, we performed exome sequencing, which revealed heterozygous mutations in the affected ITGB4 gene, c.794dupC (p. S265fs*5) and c.2962G > A (p. A988T) (Table 1). These two variants were inherited from her parents respectively. The mutation c.794dupC is a frameshift mutation that results in the early termination of the amino acid code for protein synthesis. Another mutation c.2962G > A causes the substitution of a conserved amino acid. Computer analysis predicts that this mutation may affect protein structure and function.
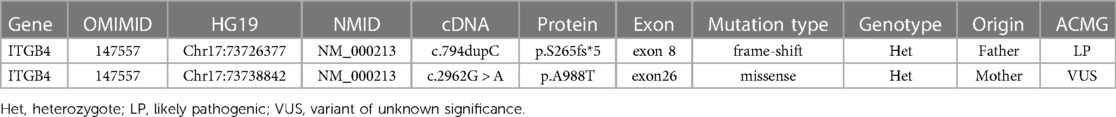
Table 1. Genotype with candidate gene abnormalities in this case.
In terms of treatment, sedation was given to reduce skin friction, and measures were taken to keep the skin clean and dry. Recombinant human epidermal growth factor gel was applied to the skin and covered with Vaseline gauze. Penicillin was also used to prevent infection. On the fourth day after admission, the baby began to develop a fever, and the result of routine blood tests showed WBC 1.89 × 109/L, Neut# 0.4 × 109/L, Hb 126 g/L, and PLT 127 × 109/L, while coagulation function revealed PT 18.7 s, APTT 70.6 s, INR 1.61. Meropenem, red blood cells and plasma were transfused, but the infection was difficult to control. The wound exudate increased and tended to bleed when touched. Abdominal distention also worsened. The patient was diagnosed with sepsis. The baby's parents refused further treatment because of the complexity of the disease, and the patient died shortly after being discharged from our hospital.
3. Literature review
Setting the search terms for pyloric atresia and epidermolysis bullosa, the subjects for 0–18-year-old patients, and the publication time from 2011 to 2021, a fuzzy search was conducted in the China National Knowledge Infrastructure (CNKI), PubMed, SinoMed, and Embase databases. According to the content of the full text and the correlation degree of EB-PA disease, 38 articles (2 Chinese articles and 36 English articles) were finally included, with a total of 49 EB-PA cases (Figure 3, Supplementary Table S1) (5–42).
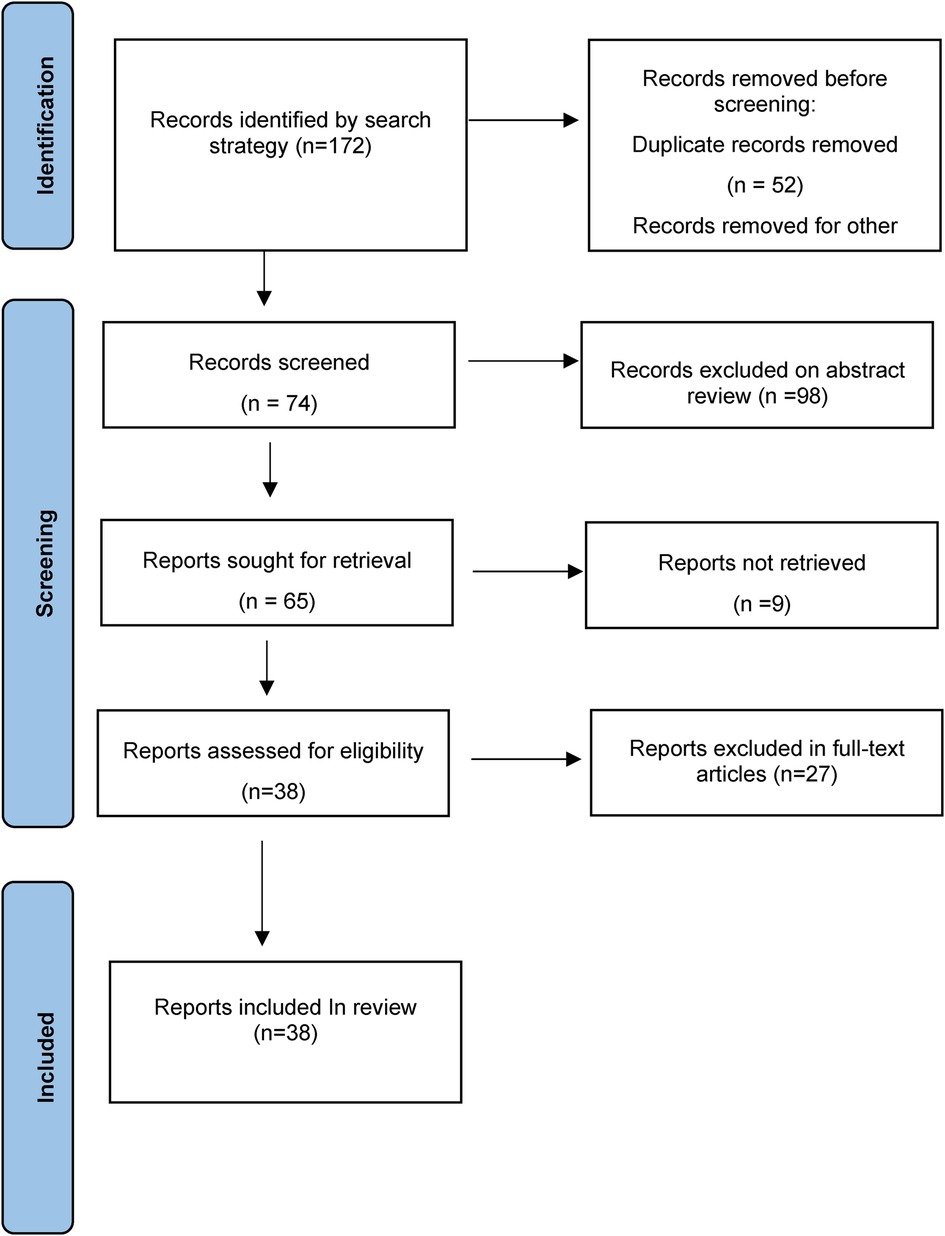
Figure 3. PRISMA flow diagram of the study selection.
3.1. Clinical characteristics
Among these 49 EB-PA cases, the gestational age at birth was documented in 46 patients, except for 3 cases whose gestational age at birth was unknown. Thirty-four were preterm, and the rest were full-term. Birth weights were reported in 28 cases, ranging from 930 to 3,640 g. A total of 28 cases recorded the results of the maternal prenatal examination, including 20 cases with hydramnios, 7 cases with upper gastrointestinal obstruction manifestations such as foetal gastric dilatation on prenatal ultrasound or foetal MRI, and 2 cases with EB typical signs such as the “snowflake sign” of amniotic fluid and chorioamniotic membrane separation on prenatal ultrasound. In addition to skin bullae and pyloric atresia, EB-PA can also be associated with other systemic malformations, especially urinary system lesions and Aplasia cutis congenita (ACC). There were 12 patients with ACC and 13 patients with urinary tract disease, mainly manifesting as polycystic kidney disease and ureter hydronephrosis. Eleven patients had hypoplasia of the outer ear, shortened nose or other abnormal facial features. Ten developed nail malnutrition.
3.2. Diagnostic methods and pathological classification
EB-PA subtypes were determined by skin biopsy, transmission electron microscopy (TEM) or immunofluorescence mapping (IFM). Thirty-two patients underwent one or more of the above examinations. Thirty-two cases of specific EB types were mentioned in the literature, including 6 cases of EBS, 25 cases of JEB and 1 case of DEB. There is also a case according to the result of immunohistochemical, is unable to judge EBS or JEB.
3.3. Molecular characteristics
Gene testing was performed in 24 patients, including 17 ITGB4 gene mutation cases. Among the ITGB4 gene mutation cases, 15 were JEB, 1 was EBS, and 1 was of an unknown type. There were 5 PLEC gene mutation cases, 4 of which were diagnosed as EBS, and the last was unclassified. Only one patient with JEB carried the ITGA6 gene mutation. Twenty-seven mutation positions of ITGB4 gene were detected, with 5 duplicated sites, c.701G > T, c.997T > G, c.3707-3725del19 and c.3793 + 1G > A, and the rest were sporadic (Supplementary Table S2). ITGB4 gene mutation sites were widely distributed, resulting in amino acid substitutions or a premature termination codon. A total of 7 PLEC gene mutation sites were detected in 4 patients (Supplementary Table S3), mainly concentrated in the exon 23–32 region, resulting in premature translation termination. Analysis of the ITGB4 gene and PLEC gene mutation sites using the gene mutation visualization program ProteinPaint (43) revealed that the ITGB4 mutation region was dominated by mutations in the integrin beta domain, with a total of 12 mutation sites located in this region, while the PLEC gene mutation position was mainly located in pletin and spectrin repeats (Figures 4, 5).
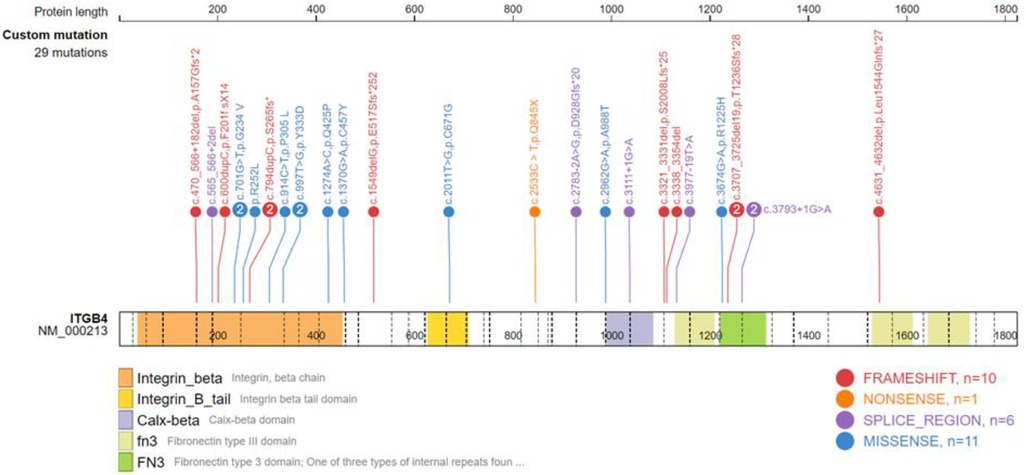
Figure 4. Schematic diagram of the ITGB4 domain and the location of the ITGB4 mutation.
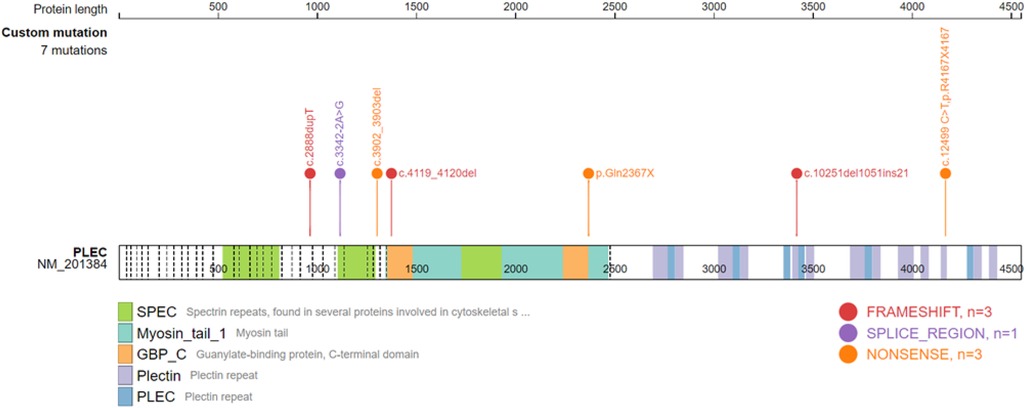
Figure 5. Schematic diagram of the PLEC domain and the location of the PLEC mutatio.
3.4. Treatment and prognosis
Forty-three patients underwent surgery for PA. Twenty-four of them died postoperatively, with the age of death varying from 5 days to 9 months, and death was related to sepsis or organ failure. Two patients abandoned treatment due to critical postoperative conditions. Five patients did not have postoperative follow-up mentioned. Twelve patients were followed up, ranging from 8 months to 36 years, and 4 of them were without symptoms. Three had mild blistering episodes, and the remainder suffered from urinary system diseases such as cystitis, hydronephrosis, or respiratory tract damage. All patients who did not receive surgical treatment died beyond a month.
4. Discussion
By summarizing the EB-PA-related cases published in the past 10 years, we found that the subtypes of EB-PA are basically EBS-PA and JEB-PA. The JEB subtype accounted for 78% of patients for whom the EB subtype had been identified. These are consistent with the international consensus. In addition, there has been a case report of DEB combined with PA (17), which is extremely rare.
Among the 49 EB-PA cases, including ours, 34 were preterm, and 22 weighed less than 2,500 g, which means that most cases of EB-PA are premature low-body weight infants. Abnormalities other than skin blisters and PA were reported in 28 patients, with urinary abnormalities being the most common malformation (13/28), followed by ACC (12/28), ear, nose and throat deformities (11/28), nail malnutrition (10/28) and respiratory tract involvement (5/28). ACC often occurs in conjunction with JEB-PA. Kayki et al. (28) speculated that ACC may be caused by the intrauterine progression of JEB-PA. Urinary system involvement of EB-PA generally manifests as renal dysplasia, such as polycystic kidney, hydronephrosis of the renal pelvis or ureter, acute tubular necrosis, urethral obstruction, ureteral cyst, kidney duplication and agenesis of the bladder (28). If the respiratory system is involved, granulation tissue formation, glottis and subhilar stenosis may arise (44). In 2010, Aydin et al. (45) first reported a child with EB-PA accompanied by partial abnormal pulmonary vein reflux (PAPVR), secondary atrial septal defect (ASD) and pulmonary hypertension, but the correlation between an abnormal circulatory system and EB-PA remains to be proven. Our case was also a low-weight preterm infant, but no concomitants other than EB-PA have been found so far.
Prenatal examination during pregnancy can help to identify a foetus with EB-PA. In our study, most of the infants showed polyhydramnios, amniotic fluid snowflake sign, or chorioamniotic membrane separation on prenatal ultrasound. The concentrations of maternal serum a-fetoprotein (AFP) and amniotic fluid AFP and acetylcholinesterase (AchE) levels are increased in some pregnant women (20, 46). Mutational analysis of fetal DNA obtained from chorionic villus sampling at 10–12 weeks of gestation or amniocentesis at 16 weeks of gestation has also been suggested (47). The postnatal diagnosis of patients with EB-PA relies on the presence of blisters, clinical manifestations of upper gastrointestinal obstruction, and various auxiliary examination methods. The definition of the subtype is necessary, mainly by immunofluorescence antigen mapping (IFM) or transmission electron microscopy (TEM) of skin biopsy tissues to identify the level of blister formation (3). However, many infants did not undergo TEM or IFM. Nine patients underwent only TEM, 10 patients underwent IFM, and 4 patients underwent both TEM and IFM.
EB-PA is commonly inherited in an autosomal recessive manner, and its pathogenesis is complex. In 1995, Vidal et al. (48) first reported that the occurrence of JEB-PA was associated with the deletion or pathogenic variation of integrin α6β4, a key protein of keratinocyte hemidesmosomes. Integrin α6β4 stabilizes the skin basement membrane by connecting laminin-332 to the dense layer beneath the basement membrane. We summarized the mutation sites of the ITGB4 gene with literature review as well as our own cases and found that 12 mutation sites were situated in the region of exons 6–15, which encode the integrin beta domain. Nine sites were located in exons 21–34, encoding the Calx-beta domain and Fibronectin type III or 3 domain. Mutations in these regions caused amino acid substitutions or premature termination of codons, resulting in reduced or absent integrin α6β4 expression. In 2005, Nakamura et al. (49) demonstrated that EBS-PA was associated with mutations in the PLEC gene located in the telomere region of the long arm of chromosome 8. Later, through gene mutation analysis, Winter et al. (50) and Charlesworth et al. (15) found that when the PLEC mutation in EBS-PA patients was located outside exon 31 or its C-terminal part, the expression of full-length and rodless plectin isoforms was significantly reduced or completely lost, thus causing EB-PA. In our study, 2 mutation sites were located in exon 32, and 1 was located in exon 31. All these mutations caused premature termination codon or splicing abnormalities. EB-PA patients with mutations in ITGA6 or PLEC genes have a high death rate. The prognosis is often poor if ITGB4 mutations are located in highly conserved or protein-binding regions (51).
The early improvement of skin lesions in patients and prevention, monitoring and treatment of complications are the main goals of the treatment of EB (52). Treatment measures for skin lesions are to avoid mechanical stress and to take good care of the skin to prevent further damage caused by skin friction. The wound can be covered with adhesive-free dressings that absorb exudate, and appropriate sedative and analgesic drugs, antihistamine receptors and other drugs can be used to reduce the symptoms of pain and itch in patients (4). In cases of secondary wound infection, antibiotics should be selected according to the results of bacterial culture and drug sensitivity tests (53). In our case, considering that the patient's lesions were too extensive and the risk of infection was high, penicillin was used prophylactically. However, on the fourth day after birth, the infection occurred despite careful care of the wound. The infection was difficult to control and eventually progressed to sepsis. In the literature review, 2 patients also died of sepsis without surgery. Hayashi et al. (54) suggested that surgical therapy of PA should be performed after the skin lesions are stabilized, which could appropriately prolong the survival time of patients. Pyloroplasty is feasible for type I and type III PA, while end-to-side or side-to-side gastroduodenostomy is required for type II PA (13). Even if PA is successfully treated by surgery, postoperative patients may die from postoperative complications such as infection, severe electrolyte imbalance, intestinal obstruction, malnutrition, and hypostatic pneumonia (5). The majority of infants in our study were treated surgically, and more than half died from complications such as sepsis, organ failure, and water-electrolyte imbalance. The postoperative mortality and complications of EB-PA were extremely high. The few surviving cases were mostly infants with mild skin lesions, but there was still a risk of repeated infection and a poor prognosis. In recent years, protein, gene or cell replacement therapy and allogeneic bone marrow transplantation have been successfully carried out in animal experiments, which are expected to be put into clinical practice (4).
In conclusion, the diagnosis and treatment of EB-PA is our current challenge. Its treatment requires multidisciplinary medical, nursing, psychological and social management (55). Even if the disease is currently improving, there is still a risk of blistering recurrence, and long-term follow-up is necessary.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
LC, YL: manuscript preparation, statistics, and manuscript writing. HZ, SY: literature search, literature analyses. YD, LY, ZM: literature analyses and reviewed the manuscript. WK: guided the project and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Guangdong Basic and Applied Basic Research Foundation of China (2019A1515011086).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2023.1098273/full#supplementary-material.
References
1. Fine J. Epidemiology of inherited epidermolysis Bullosa based on incidence and prevalence estimates from the national epidermolysis Bullosa registry. JAMA Dermatol. (2016) 152:1231. doi: 10.1001/jamadermatol.2016.2473
2. Has C, Bauer JW, Bodemer C, Bolling MC, Bruckner-Tuderman L, Diem A, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. (2020) 183:614–27. doi: 10.1111/bjd.18921
3. Fine JD, Bruckner-Tuderman L, Eady RA, Bauer EA, Bauer JW, Has C, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol. (2014) 70:1103–26. doi: 10.1016/j.jaad.2014.01.903
4. Has C, Bruckner-Tuderman L. Epidermolysis bullosa: diagnosis and therapy. Hautarzt. (2011) 62:82–90. doi: 10.1007/s00105-010-2049-x
5. Kim J, Park H, Lee H, Eom M, Choi EH. Case of epidermolysis Bullosa with pyloric atresia. Ann Dermatol. (2011) 23:S41. doi: 10.5021/ad.2011.23.S1.S41
6. Bicakci U, Tander B, Celik FC, Ariturk E, Rizalar R. Pyloric atresia associated with epidermolysis bullosa: report of two cases and review of the literature. Turkish J Trauma Emerg Surg. (2012) 18:271–3. doi: 10.5505/tjtes.2012.13284
7. Lichtenstein DA, Browne F, Daly BM, Liu L, McGrath JA, Moss C. Lethal autosomal recessive epidermolysis bullosa simplex with bowel atresia due to a novel plectin mutation. Brit J Dermatol. (2012) 167:131–2. doi: 10.1111/j.1365-2133.2012.10864.x
8. Stoevesandt J, Borozdin W, Girschick G, Hamm H, Höcht B, Kohlhase J, et al. Lethal junctional epidermolysis bullosa with pyloric atresia due to compound heterozygosity for two novel mutations in the integrin β4 gene. Klin Padiatr. (2012) 224:8–11. doi: 10.1055/s-0031-1285877
9. Diociaiuti A, Castiglia D, Morini F, Boldrini R, Fortugno P, Zambruno G, et al. Long-term follow-up of a spontaneously improving patient with junctional epidermolysis bullosa associated with ITGB4 c.3977-19T > A splicing mutation. Acta Derm Venereol. (2013) 93:116–8. doi: 10.2340/00015555-1381
10. Fu AC, Hon KL, Choi PC. A neonate with generalised bullae and pyloric atresia. Hong Kong Med J. (2013) 19:188.e1–2.23535685
11. Merrow AC, Frischer JS, Lucky AW. Pyloric atresia with epidermolysis bullosa: fetal MRI diagnosis with postnatal correlation. Pediatr Radiol. (2013) 43:1656–61. doi: 10.1007/s00247-013-2737-7
12. Mithwani AA, Hashmi A, Adil S. Epidermolysis bullosa and congenital pyloric atresia. Case Rep. (2013) 2013:bcr2013201207. doi: 10.1136/bcr-2013-201207
13. Son TN, Hoan VX. Laparoscopic management of pyloric atresia in a neonate with epidermolysis Bullosa. J Laparoendosc Adv Surg Tech. (2013) 23:649–50. doi: 10.1089/lap.2013.0189
14. Marjanovic Z, Slavkovic A, Djordjevic I. Syndromic association of pyloric atresia and epidermolysis bullosa (carmi syndrome)–a case report. West Indian Med J. (2013) 62:149–51.24564066
15. Charlesworth A, Chiaverini C, Chevrant-Breton J, DelRio M, Diociaiuti A, Dupuis RP, et al. Epidermolysis bullosa simplex with PLEC mutations: new phenotypes and new mutations. Br J Dermatol. (2013) 168:808–14. doi: 10.1111/bjd.12202
16. Hassan ME, Al Ali K, Khalaf M, Taryam L. Pyloric atresia epidermolysis bullosa aplasia cutis syndrome. Ann Pediatr Surg. (2013) 9:84–6. doi: 10.1097/01.XPS.0000428235.41499.e6
17. Short SS, Grant CN, Merianos D, Haydel D, Ford HR. A case of congenital pyloric atresia with dystrophic epidermolysis bullosa. Pediatr Surg Int. (2014) 30:681–4. doi: 10.1007/s00383-014-3505-y
18. Hon KL, Li JJ, Cheng BL, Luk DC, Murrell DF, Choi PC, et al. Age and etiology of childhood epidermolysis bullosa mortality. J Dermatolog Treat. (2015) 26:178–82. doi: 10.3109/09546634.2014.915002
19. Farmakis SG, Herman TE, Siegel MJ. Congenital pyloric atresia, type B; with junctional epidermolysis bullosa. J Perinatol. (2014) 34:572–3. doi: 10.1038/jp.2014.7
20. Dural O, Acar DK, Ekiz A, Aslan H, Polat İ, Yildirim G, et al. Prenatal ultrasound findings and a new ultrasonographic sign of epidermolysis bullosa with congenital pyloric atresia: a report of three cases. J Med Ultrason. (2014) 41:495–8. doi: 10.1007/s10396-014-0532-1
21. Joshi P, Mundada D, Shetty S, Oak S, Parelkar S, Kapadnis S, et al. Pyloric atresia-three cases and review of literature. Afr J Paediatr Surg. (2014) 11:362. doi: 10.4103/0189-6725.143178
22. Schutzman LM, Walsh M, Draus JM. Pyloric atresia: comorbidity determines outcome. J Pediatr Surg Case Rep. (2014) 2:192–5. doi: 10.1016/j.epsc.2014.03.014
23. Chahed J, Mekki M, Ksia A, Kechiche N, Hidouri S, Youssef T, et al. Management of digestive lesions associated to congenital epidermolysis bullosa. Afr J Paediatr Surg. (2015) 12:221. doi: 10.4103/0189-6725.172544
24. Yang P, Zhang Y, Chen J. Nursing care of a child with pyloric atresia complicated with epidermolysis bullosa. Chin Gen Pract Nurs. (2016) 14(31):3345–7. doi: 10.3969/j.issn.1674-4748.2016.31.045
25. Mencía Á, García M, García E, Llames S, Charlesworth A, de Lucas R, et al. Identification of two rare and novel large deletions in ITGB4 gene causing epidermolysis bullosa with pyloric atresia. Exp Dermatol. (2016) 25:269–74. doi: 10.1111/exd.12938
26. Walker GD, Woody M, Orrin E, Mellerio JE, Levy ML. Epidermolysis Bullosa with pyloric atresia and significant urologic involvement. Pediatr Dermatol. (2017) 34:e61–64. doi: 10.1111/pde.13026
27. Mitra S, Hazra S, Sengupta A. Respiratory papillomatosis in a case of carmi syndrome an unusual presentation. Int J Case Rep Images. (2017) 8:514–8. doi: 10.5348/ijcri-201773-CR-10812
28. Kayki G, Bozkaya D, Ozaltin F, Orhan D, Kaymaz F, Korkmaz E, et al. Epidermolysis Bullosa with pyloric atresia and aplasia cutis in a newborn due to homozygous mutation in ITGB4. Fetal Pediatr Pathol. (2017) 36:332–9. doi: 10.1080/15513815.2017.1324545
29. Fu C, Ma L, Chen J. Nursing care of a neonate with congenital pyloric atresia and epidermolysis bullosa. Chin J Nurs. (2017) 52(02):199–203. doi: 10.3761/j.issn.0254-1769.2017.02.016
30. Ko L, Griggs CL, Mylonas KS, Masiakos PT, Kroshinsky D. A nonlethal case of junctional epidermolysis Bullosa and congenital pyloric atresia: compound heterozygosity in a patient with a novel integrin Beta 4 gene mutation. J Pediatr. (2018) 193:261–264.e1. doi: 10.1016/j.jpeds.2017.09.023
31. Hattori M, Shimizu A, Nakano H, Ishikawa O. Mild phenotype of junctional epidermolysis bullosa with pyloric atresia due to a novel mutation of the ITGB4 gene. J Dermatol. (2018) 45:e203–204. doi: 10.1111/1346-8138.14236
32. Trah J, Has C, Hausser I, Kutzner H, Reinshagen K, Königs I. Integra®-dermal regeneration template and split-thickness skin grafting: a therapy approach to correct aplasia cutis congenita and epidermolysis Bullosa in carmi syndrome. Dermatol Ther (Heidelb). (2018) 8:313–21. doi: 10.1007/s13555-018-0237-2
33. Hicks TD, Singh H, Mikhael M, Shah AR. Carmi syndrome in a preterm neonate: a multidisciplinary approach and ethical challenge. Case Rep Pediatr. (2018) 2018:1–6. doi: 10.1155/2018/4548194
34. Al FA, Syed MK, Hussain S, Almas T, Ammar M. Carmi syndrome in a neonate: an exacting surgical challenge. Cureus. (2020) 12:e10522. doi: 10.7759/cureus.10522
35. Okulu E, Durmaz CD, Tunc G, Guzel A, Kutlay NY, Erdeve O, et al. A novel mutation in ITGB4 gene in a newborn with epidermolysis bullosa, pyloric atresia, and aplasia cutis congenita. Egypt J Med Hum Genet. (2020) 21:1–4. doi: 10.1186/s43042-020-00055-7
36. Verma KP, Robertson SJ, Winship IM. Phenotypic discordance between siblings with junctional epidermolysis bullosa-pyloric atresia. Clin Exp Dermatol. (2020) 45:793–5. doi: 10.1111/ced.14223
37. Schreiner D, Uebler A, Ginghina C, Muensterer O, Has C, Mildenberger E. Prognostic assessment and management of a patient with carmi syndrome. A case report. Int J Surg Case Rep. (2021) 84:106070. doi: 10.1016/j.ijscr.2021.106070
38. Matyas M, Miclea D, Zaharie G. Case report: uncommon association of ITGB4 and KRT10 gene mutation in a case of epidermolysis Bullosa with pyloric atresia and aplasia cutis congenita. Front Genet. (2021) 12:1–8. doi: 10.3389/fgene.2021.641977
39. Wee LWY, Tan EC, Bishnoi P, Ng YZ, Lunny DP, Lim HW, et al. Epidermolysis bullosa with pyloric atresia associated with compound heterozygous ITGB4 pathogenic variants: minimal skin involvement but severe mucocutaneous disease. Pediatr Dermatol. (2021) 38:908–12. doi: 10.1111/pde.14668
40. Chen D, Zuckerman S, Akerman Y, Shen O. A new prenatal sonographic sign of epidermolysis bullosa. J Clin Ultrasound. (2021) 49:59–61. doi: 10.1002/jcu.22922
41. Chong SC, Hon KL, Yuen LY, Choi PC, Ng WG, Chiu TW. Neonatal epidermolysis bullosa: lessons to learn about genetic counselling. J Dermatol Treat. (2021) 32:29–32. doi: 10.1080/09546634.2018.1527999
42. Ellis C, Eason C, Snyder A, Siegel M, Pai GS, Ryan E, et al. Novel missense p.R252l mutation of ITGB4 compounded with known 3793 + 1G > A mutation associated with nonlethal epidermolysis bullosa-pyloric atresia with obstructive uropathy. JAAD Case Rep. (2021) 11:63–8. doi: 10.1016/j.jdcr.2021.03.016
43. Zhou X, Edmonson MN, Wilkinson MR, Patel A, Wu G, Liu Y, et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat Genet. (2016) 48:4–6. doi: 10.1038/ng.3466
44. Ida JB, Livshitz I, Azizkhan RG, Lucky AW, Elluru RG. Upper airway complications of junctional epidermolysis Bullosa. J Pediatr. (2012) 160:657–661.e1. doi: 10.1016/j.jpeds.2011.09.029
45. Aydin M, Zenciroglu A, Yaman A, Orun UA, Arda N, Colak AG, et al. Carmi syndrome with congenital heart defects. Am J Med Genet A. (2010) 152A:2120–2. doi: 10.1002/ajmg.a.33520
46. De Jenlis Sicot B, Deruelle P, Kacet N, Vaillant C, Subtil D. Prenatal findings in epidermolysis bullosa with pyloric atresia in a family not known to be at risk. Ultrasound Obstet Gynecol. (2005) 25:607–9. doi: 10.1002/uog.1911
47. Fassihi H, McGrath JA. Prenatal diagnosis of epidermolysis bullosa. Dermatol Clin. (2010) 28:231–7, viii. doi: 10.1016/j.det.2010.02.001
48. Vidal FEA. Integrin beta 4 mutations associated with junctional epidermolysis bullosa with pyloric atresia. Nat Genet. (1995) 10:229–34. doi: 10.1038/ng0695-229
49. Nakamura H, Sawamura D, Goto M, Nakamura H, McMillan JR, Park S, et al. Epidermolysis bullosa simplex associated with pyloric atresia is a novel clinical subtype caused by mutations in the plectin gene (PLEC1). J Mol Diagn. (2005) 7:28–35. doi: 10.1016/S1525-1578(10)60005-0
50. Winter L, Wiche G. The many faces of plectin and plectinopathies: pathology and mechanisms. Acta Neuropathol. (2013) 125:77–93. doi: 10.1007/s00401-012-1026-0
51. Mylonas KS, Hayes M, Ko LN, Griggs CL, Kroshinsky D, Masiakos PT. Clinical outcomes and molecular profile of patients with carmi syndrome: a systematic review and evidence quality assessment. J Pediatr Surg. (2019) 54:1351–8. doi: 10.1016/j.jpedsurg.2018.05.019
52. Sprecher E. Epidermolysis bullosa simplex. Dermatol Clin. (2010) 28:23–32. doi: 10.1016/j.det.2009.10.003
53. El HM, Zambruno G, Bourdon-Lanoy E, Ciasulli A, Buisson C, Hadj-Rabia S, et al. Multicentre consensus recommendations for skin care in inherited epidermolysis bullosa. Orphanet J Rare Dis. (2014) 9:76. doi: 10.1186/1750-1172-9-76
54. Hayashi AH, Galliani CA, Gillis DA. Congenital pyloric atresia and junctional epidermolysis bullosa: a report of long-term survival and a review of the literature. J Pediatr Surg. (1991) 26:1341. doi: 10.1016/0022-3468(91)90616-2
Keywords: epidermolysis bullosa, pyloric atresia, newborns, molecular characteristics, clinical characteristics, exome sequencing
Citation: Luo C, Yang L, Huang Z, Su Y, Lu Y, Yu D, Zhang M and Wu K (2023) Case report: A case of epidermolysis bullosa complicated with pyloric atresia and a literature review. Front. Pediatr. 11:1098273. doi: 10.3389/fped.2023.1098273
Received: 14 November 2022; Accepted: 9 March 2023;
Published: 23 March 2023.
Edited by:
Melinda Matyas, University of Medicine and Pharmacy Iuliu Hatieganu, RomaniaReviewed by:
Hisayoshi Kawahara, Naramachi Hospital, JapanBurak Tander, Acıbadem University, Türkiye
© 2023 Luo, Yang, Huang, Su, Lu, Yu, Zhang and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kai Wu d3VrYWlAc211LmVkdS5jbg==
Specialty Section: This article was submitted to Genetics of Common and Rare Diseases, a section of the journal Frontiers in Pediatrics