 Oksana Boyarchuk
Oksana Boyarchuk Olha Dyvonyak2
Olha Dyvonyak2 Alla Volokha
Alla Volokha- 1Department of Children's Diseases and Pediatric Surgery, I.Horbachevsky Ternopil National Medical University, Ternopil, Ukraine
- 2Municipal Children's Hospital, Ternopil, Ukraine
- 3Department of Pediatrics, Pediatric Infectious Diseases, Immunology and Allergology, Shupyk National Healthcare University of Ukraine, Kyiv, Ukraine
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), also known as autoimmune polyglandular syndrome type 1 (APS-1) is a rare autosomal recessive inborn error of immunity (IEI), which is accompanied by immune dysregulation. Hypoparathyroidism, adrenocortical failure and candidiasis are its typical manifestations. Here we report about recurrent COVID-19 in a 3-year-old boy with APECED, who developed retinopathy with macular atrophy and autoimmune hepatitis after the first episode of SARS-CoV-2 infection. Primary Epstein-Barr virus infection and a new episode of SARS-CoV-2 infection with COVID pneumonia triggered the development of severe hyperinflammation with signs of hemophagocytic lymphohistiocytosis (HLH): progressive cytopenia (thrombocytopenia, anemia, lymphopenia), hypoproteinemia, hypoalbuminemia, high levels of liver enzymes, hyperferritinemia, increased triglycerides levels; and coagulopathy with a low level of fibrinogen. Treatment with corticosteroids and intravenous immunoglobulins did not lead to a significant improvement. The progression of HLH and COVID-pneumonia resulted in a fatal outcome. The rarity and varied presentation of the HLH symptoms led to diagnostic difficulties and diagnosis delay. HLH should be suspected in a patient with immune dysregulation and impaired viral response. Treatment of infection-HLH is a major challenge due to the difficulties in balancing immunosuppression and management of underlying/triggering infection.
Introduction
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), also known as autoimmune polyglandular syndrome type 1 (APS-1) is a rare autosomal recessive inborn error of immunity (IEI), which is accompanied by immune dysregulation (1, 2). APECED is caused by a mutation in the Autoimmune Regulator (AIRE) gene and characterized by chronic mucocutaneous candidiasis (CMC) and multisystem autoimmunity. Other frequently reported symptoms include hypoparathyroidism and adrenocortical failure (1), and together with candidiasis they form a classic triad (2). The presence of at least two signs of the triad is required for diagnosis.
Other organs and systems can be affected with various frequency and these most often involve the endocrine organs and autoimmunity, such as hypothyroidism, type 1 diabetes mellitus, pernicious anemia, alopecia, vitiligo, malabsorption, Sjogren-like syndrome, pneumonitis, and hepatitis (2–5).
Candidiasis of the skin or mucous membranes is usually one of the first symptoms; most frequently it presents as a recurrent oral candidiasis or diaper dermatitis (6). The frequency of APECED-associated hepatitis ranges from about 10% in the European population to 42% in American cohorts (2), and in a third of the patients in the American cohort, hepatitis symptoms preceded the other classic APECED criteria.
Ocular manifestations are also reported in patients with APECED and are associated with a poor prognosis (6–9). Keratopathy and retinopathy are the most frequent among ocular abnormalities (6–8). Vision loss due to macular atrophy had also been detected. Specific autoantibodies against corneal and/or retinal antigens are the main causes responsible for the ocular manifestations in patients with APECED (8). Moreover, there is no correspondence between the severity of eye symptoms and other systemic manifestations.
Besides CMC, patients with APECED have a high susceptibility to some other specific infections that are mediated by anti-cytokine autoantibodies and/or Tcell driven autoimmune tissue injury (10). High titers of autoantibodies to IFN-α present in almost every patient with APECED result the severe course of viral infections in this cohort of patients (10, 11). This has become especially relevant during the COVID-19 pandemic, because the presence of autoantibodies to type I IFNs underlies the severe COVID-19 pneumonia in patients with APECED (12–14).
Here we report about recurrent COVID-19 in a 3-year-old boy with APECED, who developed retinopathy with macular atrophy and autoimmune hepatitis after the first episode of SARS-CoV-2 infection. Primary Epstein-Barr virus (EBV) infection and a new episode of SARS-CoV-2 infection with COVID pneumonia triggered the development of severe hyperinflammation with signs of hemophagocytic lymphohistiocytosis (HLH), which led to a fatal outcome.
Case report
A 3-year-8-month-old boy was admitted to the hospital in January, 2022 with complaints of fever up to 39°C, runny nose, cough, reduced appetite, irritability.
The first symptoms appeared 5 days before the admission. He was treated for an upper respiratory infection, received symptomatic and antibiotic therapy. His condition did not improve, leading to his hospitalization.
The patient's history showed that he was the first child of non-consanguineous parents. His weight at birth was 3,080 g. Vaccinations were carried out according to the schedule. On the 11th day after the first measles, mumps, and rubella (MMR) vaccination, the boy developed symptoms of measles (fever, rash), for which he was hospitalized.
In September 2020, the boy had COVID-19 after a contact with sick family members. The course of the disease was mild, accompanied by a cough for several days. However, a few days after COVID-19, the mother noticed a rapid progressive deterioration of the child's vision, up to a complete blindness. The boy was examined for this complaint in several clinics and was diagnosed with retinopathy and secondary macular dystrophy of unknown origin. During a hospitalization in November 2020, additional tests to clarify the diagnosis revealed acute otitis media, candidiasis of the oral mucosa and an increase in the levels of liver enzymes: aspartate aminotransferase (AST) up to 228.6 U/L, alanine aminotransferase (ALT) up to 566.5 U/L, and lactate dehydrogenase (LDH) up to 376.6 U/L (see Table 2 for the reference ranges). The levels of bilirubin, alkaline phosphatase, albumin, gamma-glutamyl transferase (GGT) were within normal limits. Leukocytosis of 15.7 109/L was detected, while other values of the complete blood count (CBC), including the level of platelets, as well as the coagulogram indexes were within normal limits. Tests were carried out to exclude viral hepatitis B and C, cytomegalovirus (CMV) infection, Epstein-Barr virus (EBV) infection, toxoplasmosis, rubella, and HIV infection. IgM and/or IgG antibodies to the indicated pathogens were negative, except antibodies to rubella (high titer of IgG to rubella antibodies), which could indicate that the patient had rubella or the presence of post-vaccination antibodies.
Given the development of measles after MMR vaccination, the signs of liver damage, and moderate manifestations of candidiasis, a congenital pathology, such as inborn errors of immunity, was suspected. Immunological examination revealed no significant abnormalities, except for a slightly increased level of IgG (Table 1). Whole exome sequencing (WES) detected a mutation in the AIRE gene, which was confirmed by gene sequencing. Two homozygous pathogenic variants, c.769C > T (p.Arg257 Ter), were identified in AIRE. Among the markers of autoimmune hepatitis, only liver cytosolic antigen (LC-1, IgG antigen) was positive.
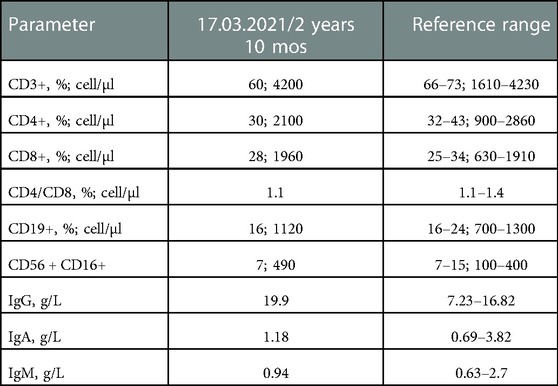
Table 1. Immunological parameters of the patient.
Autoimmune hepatitis and oropharyngeal candidiasis were diagnosed as signs of APECED. The patient received azathioprine, oral glucocorticoids with a gradual dose reduction and subsequent withdrawal, antifungal therapy, calcium and vitamin D. On the background of treatment, the levels of liver enzymes have normalized. Glucocorticoids were discontinued and the patient continued azathioprine intake.
In admission to the hospital in January 2022, the patient's condition was estimated as moderate. His weight was 15 kg (z −0.32), height 94 cm (z −1.36). The child was irritable. Slight swellings on the face, cracks on the lips were observed. The throat was hyperemic. The heart rate was 130 beats per minute, respiratory rate - 26 breaths per minute, and oxygen saturation - 96%–98%. Vesicular breathing was heard over the thorax. The liver protruded 2 cm from the edge of the right costal arch, the spleen was located along the edge of the left costal arch.
Considering prolonged fever, hyperemic throat, increased incidence of SARS-CoV-2 infection, associated with the Omicron variant COVID-19 was suspected, but the PCR test for COVID-19 was negative on admission. CBC revealed leukopenia, moderate lymphopenia, thrombocytopenia, a pronounced shift of the indices to the left (33% of band neutrophils), and elevated erythrocyte sedimentation rate (ESR) (Table 2). Biochemical tests showed hypoproteinemia, hypoalbuminemia, hyperbilirubinemia due to direct bilirubin, high levels of ALT, AST, GGT, LDG, and a reduced calcium level (Table 2). CRP was elevated (50.3 mg/L), while procalcitonin was normal (0.13 ng/ml). A high level of ferritin was detected (1,297 ng/ml). Chest x-ray on admission showed no pathological changes. Echocardiographic findings did not reveal cardiac dysfunction, pericarditis, valvulitis, or coronary abnormalities.
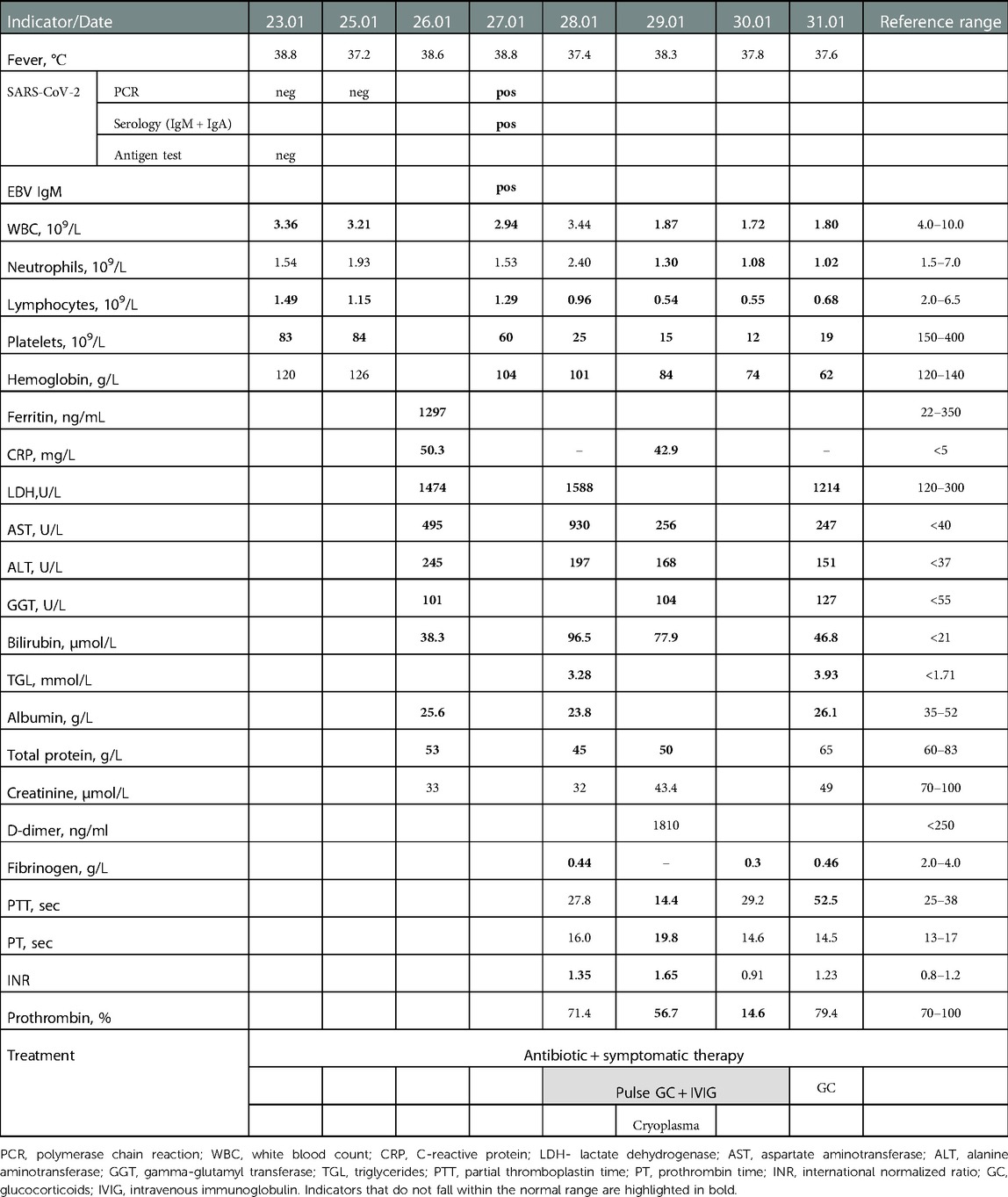
Table 2. Clinical data, laboratory test results and treatment of the patient.
Given the negative PCR test for COVID-19, an examination was conducted to identify other causes of hepatitis. Hepatitis B and C markers (HBsAg, anti HCV IgM) were negative. However, a high titer of IgM antibodies to the Epstein-Barr virus (55.68) was observed with a normal IgG indicator to the Epstein-Barr virus, 3.66 (reference <9). On the 4th day after admission (9th day after the onset of symptoms), a positive serology result for СОVID-19 was obtained: IgM + IgA antibodies to SARS-CoV-2 were positive (64.38; reference range 6–8); positive PCR test for COVID-19 was also obtained.
Prescribed symptomatic treatment and antibiotic therapy did not improve the patient's condition. The fever was maintained, oral candidiasis appeared. Hepatosplenomegaly increased (liver + 4 cm, spleen + 2 cm). Oxygen saturation decreased to 90%–92%.
Multisystem inflammatory syndrome in children (MIS-C), associated with COVID-19 was suspected considering hyperinflammation that presented as fever and elevated markers of inflammation (CRP, ESR, ferritin, LDH, lymphocytopenia, hypoalbuminemia).
However, lack of clinical signs of multisystem involvement according to WHO case definition of MIS-C (15), coagulopathy with a low level of fibrinogen, normal procalcitonin, the absence of neutrophilia and the development of neutropenia, increased triglycerides levels, alternative plausible diagnoses listed in the CDC case definition of MIS-C (16) more support to viral related secondary HLH or macrophage activation syndrome (MAS).
On the 5th day, due to the lack of positive dynamics and worsening symptoms, the child was transferred to the intensive care unit. Oxygen therapy, intravenous immunoglobulin (2 g/kg over 3 days), intravenous pulse methylprednisolone (10 mg/kg/day N 3), cryoplasma (10 ml/kg/day), and antifungal therapy (fluconazole 6 mg/kg/day) were prescribed, and antibiotic therapy (cefepime 100 mg/kg/day) was continued.
Despite the treatment, slight positive dynamics were observed only in some biochemical indicators (bilirubin, ALT, AST) and indicators of the coagulogram (Table 2). However, anemia, thrombocytopenia, leukopenia, and lymphopenia have exacerbated, and hypofibrinogenemia persisted (Table 2). On repeated chest x-ray, bilateral polysegmental pneumonia and right-sided exudative pleurisy were detected. Antibiotic therapy was changed to meropenem (60 mg/kg/day), additional cryoplasma (10 ml/kg/day), was prescribed. The boy was placed on a ventilator, but the escalating multiple organ failure led to the child's death.
Discussion
The peculiarity of the presented case is a reсurrent COVID-19 infection complicated by HLH with a fatal outcome in an immunocompromised child with APECED. The first episode of COVID-19 in September 2020 had a mild course, but led to the manifestation of the symptoms of IEI. While these symptoms (retinopathy, hepatitis), except for the moderate mucosal candidiasis, are not included in the triad of classic APECED symptoms, taken together with the reaction to the live vaccine, it was possible to suspect immunodeficiency.
Immunosuppressive therapy led to stabilization of the hepatitis, but the eye symptoms had no positive dynamics. A second episode of COVID-19 occurred in January 2022. The course presented with prolonged, treatment resistant fever for 2 weeks, progressive cytopenia (thrombocytopenia, anemia, lymphopenia), hypoproteinemia, hypoalbuminemia, signs of active hepatitis (hyperbilirubinemia due to the direct fraction, high levels of liver enzymes), hyperferritinemia, increased triglycerides level; coagulopathy with a low level of fibrinogen. It was only possible to confirm COVID-19 on the 9th day after the onset of symptoms. Later, the signs of pneumonia arose, which was confirmed by x-ray. The patient met some criteria of MIS-C, related to SARS-CoV-2 infection, however, a low level of fibrinogen, the absence of neutrophilia and the development of neutropenia, increased triglycerides levels were more consistent with viral related secondary HLH.
Another feature of current case is the presence of a mixed infection: an active EBV infection was detected (by high positive IgM antibodies) along with COVID-19. The absence of IgG to EBV may indicate a primary infection that affected the course of COVID-19 and was also a likely trigger for hyperinflammation and the development of HLH/MAS.
In general, COVID-19 in patients with APECED has a severe course, due to the presence of antibodies to type I IFN in such patients, which has been confirmed by several recent studies (10–14). Autoantibodies to type I IFNs affect the severity of the course of COVID-19 in the general population and account for ∼20% of COVID-19 deaths (17). Neutralizing autoantibodies to type-I IFNs in patients with critical COVID-19 pneumonia are associated with delayed time to viral clearance (18). Unfortunately, we could not determine the presence of antibodies to type I IFN in our patient, although other studies indicate a high titer of them in nearly 100% of patients with APECED (11, 12). The involvement of other bronchial antigens, such as BPIFB1 and potassium regulator KCNRG was confirmed in the development of autoimmune pneumonitis in patients with APECED and some of lung epithelial self-antigens remain undetected (10, 19).
The other potential mechanism of severe COVID-19 in patients with APECED may be associated with impaired negative selection of autoreactive T-cells in connection with AIRE deficiency (10). These T-cells infiltrate different organs, including lungs and spleen, causing organ damage and dysfunction, and predisposing to secondary invasive bacterial infections (10).
In our opinion, the cause of death in the presented case was not only COVID pneumonia, but to a large extent also HLH. The pathophysiological basis of HLH/MAS is pathological immune activation, or the so-called cytokine storm (20, 21). HLH/MAS is characterized by fever, variable hepatosplenomegaly, high levels of CRP and ferritin, hemophagocytosis, cytopenias (including pancytopenia), and coagulopathy associated with liver pathology and disseminated intravascular coagulation (20). Initially, the MAS terminology was used in rheumatology regarding systemic juvenile idiopathic arthritis (sJIA) and adult-onset Still disease. It also corresponds to the secondary (acquired) HLH in autoimmune, oncological diseases or in combination with infections or other diseases (20), whereas primary HLH was defined as a pediatric monogenic immunodeficiency state with hyperinflammation (20).
Viral infections are the most common triggers of HLH/MAS in children (20–22). Among them, EBV is the most frequently identified, followed by CMV (17–19). However, other bacterial, parasitic, and fungal infections can also trigger HLH/MAS (20, 21).
The issue of cytokine storm and HLH became very relevant during the COVID-19 pandemic. Hyperferritinemia and coagulopathy associated with SARS-CoV-2 infection have been linked to a HLH -type phenotype, but the incidence of classical HLH/MAS in patients with COVID-19 is not well described (20).
HLH also occurs in various groups of immunocompromised patients, including those with IEI (21). Patients with severe combined immunodeficiency, Omenns syndrome, severe DiGeorge syndrome, Wiskott-Aldrich syndrome, X-linked agammaglobulinemia, chronic granulomatous disease, autoimmune lymphoproliferative syndrome have all been reported to present with HLH (23). Patients with IEI complicated by HLH often have severe, unresolved infections (21, 24). To the best of our knowledge, the reported case is the first patient with APECED who developed HLH. Unfortunately, combined HLH and COVID pneumonia have caused the patient's death.
The development of hyperinflammation in COVID-19 pneumonia can mimic signs of HLH. McGonagle et al. (20) point out the main differences between hyperinflammation in COVID-19 and MAS. With COVID-19 pneumonia, there is a localized tissue-specific cytokine response with the development of “local cytokine flooding”, while MAS is characterized by systemic macrophage activation and a global cytokine storm (20). In addition, concentrations of cytokines and ferritins in patients with COVID-19 pneumonia are lower than in patients with MAS.
In this case the potential presence of autoantibodies to type I IFNs might impair immune response, which led to a massive cytokine release syndrome with the development of systemic hyperinflammation. Impaired negative selection of autoreactive T-cells may have also contributed to cytokine storm and organs injury. Prolonged viral infection and antigenic stimulation led to the activation of T cells and macrophages, as well as the massive release of pro-inflammatory cytokines (25).
The HLH 2004 (26) and MAS 2016 (27) diagnostic criteria are summarized in Table 3. Symptoms of the patient fully correspond to these criteria, including 5 out of 8 criteria of HLH 2004 in addition to the presence of fever, hyperferritinemia and all other 4 criteria of MAS.
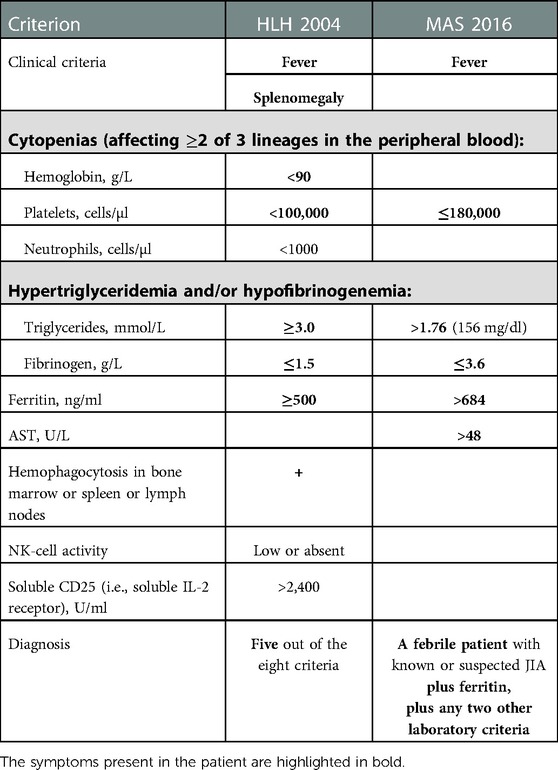
Table 3. Compliance of the patient's symptoms with the classification criteria for HLH 2004 and MAS 2016.
HLH/MAS treatment also remains a challenging issue. Immune dysregulation is the central problem in both APECED and HLH, therefore immunosuppressive therapy should be the priority (20, 21). The patient received pulse therapy with methylprednisolone and intravenous immunoglobulins in a suppressive dose, but this did not lead to a significant improvement of his condition. Anti-cytokine therapies, including anti-interleukin-1 (IL-1), IL-6, IL-18, interferon-γ, and janus kinase (JAK) could have been used as the second-line drugs in the treatment of the patient (20, 25, 28). Another drug has been reported to improve the clinical symptoms of the disease as a part of a complex treatment of Epstein Barr virus induced HLH is rituximab (29).
The verifiable presence of autoantibodies to type I IFNs in patients with APECED also requires the prescription of immunosuppressive therapy or other strategies to overcome the effect of autoantibodies to type I IFNs (12–14).
The other strategy is supporting immune response against SARS-CoV-2 virus using monoclonal antibodies to the SARS-CoV-2 spike protein (14), which block entry of SARS-CoV-2 in host cells Prescription of bamlanivimab and etesevimab for two siblings with APECED allowed to prevent invasive ventilatory support, admission to the intensive care, and death of these patients (14).
Thus, the initial symptoms of HLH/MAS are nonspecific and may overlap with other infectious, inflammatory and oncohematology conditions (21), especially in the COVID-19 era. Hyperinflammation in patients with COVID-19, including MIS-C can mimic and overlap with the signs of HLH. Rarity and diversity of the HLH/MAS symptoms led to diagnostic difficulties and diagnosis delay (21). Patients with IEI, especially with immune dysregulation are at high risk of HLH.
Rising awareness of symptoms of IEI, cytokine storm and HLH/MAS should improve their recognition (21, 30, 31) and accelerate the administration of appropriate treatment. However, some authors (21) alert about the risk of misdiagnosis and inappropriate treatment of HLH-mimicking conditions.
The limitation of this study is the impossibility to establish a definitive cause-effect relationship. The lack of certain examinations (EBV-DNA, cytokine levels) and the post-mortem diagnosis did not allow to fully determine the degree of damage to different organs and tissues. The short period between the diagnosis and death of the child did not permit to use the entire spectrum of treatment possibilities.
Thus, HLH should be suspected in a patient with immune dysregulation and impaired viral response. Treatment of infection-HLH is a major challenge due to the difficulties in balancing immunosuppression and management of underlying/triggering infection.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Ethical Committee of I.Horbachevsky Ternopil National Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
OB designed and conceptualised the manuscript, collected data and drafted the initial manuscript. OD conducting clinical research and patient care, reviewed and revised the manuscript. TH collected data, reviewed and revised the manuscript. AV designed and conceptualised the manuscript, reviewed and revised the manuscript. All authors read, critically reviewed and approved the final version. All authors contributed to the article and approved the submitted version.
Acknowledgments
We would like to thank the doctors of Ternopil Municipal Children's Hospital, Ternopil Regional Children's Hospital and Western-Ukrainian Specialized Children's Medical Center, Lviv, Ukraine (Larysa Kostyuchenko, Vita Voloshchuk) for their collaboration.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Valenzise M, Alessi L, Bruno E, Cama V, Costanzo D, Genovese C, et al. APECED Syndrome in childhood: clinical spectrum is enlarging. Minerva Pediatr. (2016) 68(3):226–9. PMID: 25502918
2. Chascsa DM, Ferré EMN, Hadjiyannis Y, Alao H, Natarajan M, Quinones M, et al. APECED-Associated Hepatitis: clinical, biochemical, histological and treatment data from a large, predominantly American cohort. Hepatology. (2021) 73(3):1088–104. doi: 10.1002/hep.31421
3. Kluger N, Jokinen M, Krohn K, Ranki A. Gastrointestinal manifestations in APECED syndrome. J Clin Gastroenterol. (2013) 47:112–20. doi: 10.1097/MCG.0b013e31827356e1
4. Ferre EMN, Break TJ, Burbelo PD, Allgäuer M, Kleiner DE, Jin D, et al. Lymphocyte-driven regional immunopathology in pneumonitis caused by impaired central immune tolerance. Sci Transl Med. (2019) 11:eaav5597. doi: 10.1126/scitranslmed.aav5597
5. Sakaguchi H, Mizuochi T, Haruta M, Takase R, Yoshida S, Yamashita Y, et al. AIRE Gene mutation presenting at age 2 years with autoimmune retinopathy and steroid-responsive acute liver failure: a case report and literature review. Front Immunol. (2021) 12:687280. doi: 10.3389/fimmu.2021.687280
6. Ma Y, Wang X, Li R. AIRE Gene mutation predisposing chronic mucocutaneous candidiasis and pigmented retinitis in two kids from a Chinese family. Emerg Microbes Infect. (2022) 11(1):1705–6. doi: 10.1080/22221751.2022.2090860
7. Guo CJ, Leung PSC, Zhang W, Ma X, Gershwin ME. The immunobiology and clinical features of type 1 autoimmune polyglandular syndrome (aps-1). Autoimmun Rev. (2018) 17(1):78–85. doi: 10.1016/j.autrev.2017.11.01
8. Couturier A, Brézin AP. Ocular manifestations of autoimmune polyendocrinopathy syndrome type 1. Curr Opin Ophthalmol. (2016) 27(6):505–13. doi: 10.1097/ICU.0000000000000306
9. Bourgault S, Baril C, Vincent A, Héon E, Ali A, MacDonald I, et al. Retinal degeneration in autoimmune polyglandular syndrome type 1: a case series. Br J Ophthalmol. (2015) 99(11):1536–42. doi: 10.1136/bjophthalmol-2014-305897
10. Oikonomou V, Break TJ, Gaffen SL, Moutsopoulos NM, Lionakis MS. Infections in the monogenic autoimmune syndrome APECED. Curr Opin Immunol. (2021) 72:286–97. doi: 10.1016/j.coi.2021.07.011
11. Kisand K, Link M, Wolff AS, Meager A, Tserel L, Org T, et al. Interferon autoantibodies associated with AIRE deficiency decrease the expression of IFN-stimulated genes. Blood. (2008) 112(7):2657–66. doi: 10.1182/blood-2008-03-144634
12. Bastard P, Orlova E, Sozaeva L, Lévy R, James A, Schmitt MM, et al. Preexisting autoantibodies to type I IFNs underlie critical COVID-19 pneumonia in patients with APS-1. J Exp Med. (2021) 218(7):e20210554. doi: 10.1084/jem.20210554
13. Beccuti G, Ghizzoni L, Cambria V, Codullo V, Sacchi P, Lovati E, et al. A COVID-19 pneumonia case report of autoimmune polyendocrine syndrome type 1 in lombardy, Italy: letter to the editor. J Endocrinol Invest. (2020) 43(8):1175–7. doi: 10.1007/s40618-020-01323-4
14. Ferré EMN, Schmitt MM, Ochoa S, Rosen LB, Shaw ER, Burbelo PD, et al. SARS-CoV-2 spike protein-directed monoclonal antibodies may ameliorate COVID-19 complications in APECED patients. Front Immunol. (2021) 12:720205. doi: 10.3389/fimmu.2021.720205
15. World Health Organization. Multisystem inflammatory syndrome in children and adolescents with COVID-19 (2020). Available at: https://www.who.int/publications/i/item/multisystem-inflammatory-syndrome-in-children-and-adolescents-with-COVID-19
16. Centers for Disease Control and Prevention. Emergency preparedness and response: health alert network (2020). Available at: https://emergency.cdc.gov/han/2020/han00432.asp
17. Bastard P, Gervais A, Le Voyer T, Rosain J, Philippot Q, Manry J, et al. Autoantibodies neutralizing type I IFNs are present in ∼4% of uninfected individuals over 70 years old and account for ∼20% of COVID-19 deaths. Sci Immunol. (2021) 6(62):eabl4340. doi: 10.1126/sciimmunol.abl4340
18. Abers MS, Rosen LB, Delmonte OM, Shaw E, Bastard P, Imberti L, et al. Neutralizing type-I interferon autoantibodies are associated with delayed viral clearance and intensive care unit admission in patients with COVID-19. Immunol Cell Biol. (2021) 99(9):917–21. doi: 10.1111/imcb.12495
19. Alimohammadi M, Dubois N, Sköldberg F, Hallgren A, Tardivel I, Hedstrand H, et al. Pulmonary autoimmunity as a feature of autoimmune polyendocrine syndrome type 1 and identification of KCNRG as a bronchial autoantigen. Proc Natl Acad Sci U S A. (2009) 106(11):4396–401. doi: 10.1073/pnas.0809986106
20. McGonagle D, Ramanan AV, Bridgewood C. Immune cartography of macrophage activation syndrome in the COVID-19 era. Nat Rev Rheumatol. (2021) 17(3):145–57. doi: 10.1038/s41584-020-00571-1
21. Jordan MB, Allen CE, Greenberg J, Henry M, Hermiston ML, Kumar A, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: recommendations from the north American consortium for histiocytosis (NACHO). Pediatr Blood Cancer. (2019) 66(11):e27929. doi: 10.1002/pbc.27929
22. Fitzgerald MP, Armstrong L, Hague R, Russell RK. A case of EBV driven haemophagocytic lymphohistiocytosis complicating a teenage Crohn's Disease patient on azathioprine, successfully treated with rituximab. J Crohns Colitis. (2013) 7(4):314–7. doi: 10.1016/j.crohns.2012.05.002
23. Xu XJ, Wang HS, Ju XL, Xiao PF, Xiao Y, Xue HM, et al. Clinical presentation and outcome of pediatric patients with hemophagocytic lymphohistiocytosis in China: a retrospective multicenter study. Pediatr Blood Cancer. (2017) 64(4):e26264. doi: 10.1002/pbc.26264
24. Bode SF, Ammann S, Al-Herz W, Bataneant M, Dvorak CC, Gehring S, et al. The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis. Haematologica. (2015) 100(7):978–88. doi: 10.3324/haematol.2014.121608
25. Henderson LA, Cron RQ. Macrophage activation syndrome and secondary hemophagocytic lymphohistiocytosis in childhood inflammatory disorders: diagnosis and management. Paediatr Drugs. (2020) 22(1):29–44. doi: 10.1007/s40272-019-00367-1
26. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. (2007) 48(2):124–31. doi: 10.1002/pbc.21039
27. Ravelli A, Minoia F, Davì S, Horne A, Bovis F, Pistorio A, et al. 2016 Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a European league against rheumatism/American college of rheumatology/paediatric rheumatology international trials organisation collaborative initiative. Arthritis Rheumatol. (2016) 68:566–76. doi: 10.1002/art.39332
28. Frigault MJ, Nikiforow S, Mansour MK, Hu ZH, Horowitz MM, Riches ML, et al. Tocilizumab not associated with increased infection risk after CAR T-cell therapy: implications for COVID-19? Blood. (2020) 136(1):137–9. doi: 10.1182/blood.2020006216
29. Chellapandian D, Das R, Zelley K, Wiener SJ, Zhao H, Teachey DT, et al. Treatment of epstein barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemoimmunotherapeutic regimens. Br J Haematol. (2013) 162(3):376–82. doi: 10.1111/bjh.12386
30. Boyarchuk O, Volyanska L, Kosovska T, Lewandowicz-Uszynska A, Kinash M. Awareness of primary immunodeficiency diseases among medical students. Georgian Med News. (2018) 285:124–30.
Keywords: AIRE, APECED, APS-1, COVID-19, macrophage activation syndrome, haemophagocytic lymphohistiocytosis
Citation: Boyarchuk O, Dyvonyak O, Hariyan T and Volokha A (2023) Case report: Virus-induced hemophagocytic lymphohistiocytosis in a patient with APECED. Front. Pediatr. 11:1086867. doi: 10.3389/fped.2023.1086867
Received: 1 November 2022; Accepted: 30 January 2023;
Published: 15 February 2023.
Edited by:
Alenka Gagro, Children's Hospital Zagreb, CroatiaReviewed by:
Francesca Minoia, IRCCS Ca 'Granda Foundation Maggiore Policlinico Hospital, ItalyAnna Sediva, University Hospital in Motol, Czechia
Angel Robles-Marhuenda, University Hospital La Paz, Spain
© 2023 Boyarchuk, Dyvonyak, Hariyan and Volokha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oksana Boyarchuk Ym95YXJjaHVrQHRkbXUuZWR1LnVh
Specialty Section: This article was submitted to Pediatric Immunology, a section of the journal Frontiers in Pediatrics