
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 01 March 2023
Sec. Pediatric Hematology and Hematological Malignancies
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1067131
This article is part of the Research Topic Case Reports in Pediatric Hematology and Hematological Malignancies 2022 View all 14 articles
 Abraham Ipe1*
Abraham Ipe1* Anne Angiolillo1,2David Jacobsohn1,3
Anne Angiolillo1,2David Jacobsohn1,3 Jinjun Cheng1,4
Jinjun Cheng1,4 Miriam Bornhorst1,5
Miriam Bornhorst1,5 Joyce Turner1,5
Joyce Turner1,5 Anant Vatsayan1,3
Anant Vatsayan1,3
Background: Germline Checkpoint Kinase 2 gene (CHEK2) mutations can increase the risk of solid tumors. Recently, they have been identified as risk factors for hematologic malignancies. However, to the best of our knowledge, B-acute lymphoblastic leukemia (B-ALL) has never been described as a presenting manifestation of germline CHEK2 mutation. Chimeric antigen receptor-T (CAR-T) cell therapy directed against CD19 antigen (tisagenlecleucel) is a novel cellular therapy for treatment of relapsed/refractory (R/R) B-ALL. The use of tisagenlecleucel has not been described in patients with CHEK2 mutation.
Case Presentation: We describe a case of a pediatric patient with a heterozygous pathogenic germline CHEK2 mutation (c.1100delC; p.Thr367Metfs*15) successfully treated with tisagenlecleucel for relapsed B-ALL to avoid hematopoietic cell transplant (HCT). The twelve-year-old boy was diagnosed with National Cancer Institute (NCI) high-risk B-ALL (white blood cell count >50,000/mcL), with no extramedullary disease. Cytogenetic analysis revealed normal karyotype but fluorescent in situ hybridization (FISH) showed 93% positivity for CRLF2::P2RY8 rearrangement. He was treated as per Children's Oncology Group (COG) AALL1131 therapy and achieved a complete remission. Seven months after diagnosis, he was found to have papillary thyroid carcinoma with no evidence of metastatic disease. The patient underwent a total thyroidectomy with central lymph node biopsy and radioactive iodine therapy. The patient's biological mother and fraternal twin brother carry the same germline CHEK2 mutation with no history of malignancy. The biological father tested negative for the familial mutation. The patient's genetic panel also identified three variants of unclear significance: CDKN2A (c.37 °C > T; p.Arg124Cys), FLCN (c.62G > A; p.Cys21Tyr) and SDHAF2 (c.139A > G; p.Met47Val). Extended family history also revealed a diagnosis of anaplastic thyroid cancer in maternal uncle at the age of 44 years. Fifteen months after diagnosis the patient had a relapse of B-ALL (both medullary and extramedullary with blasts in CSF), which was successfully treated with tisagenlecleucel. The patient remains in remission 3 years after receiving tisagenlecleucel.
Conclusion: As conventional chemotherapy and radiation can potentially increase the risk of DNA damage and development of secondary malignancies, CD19 CAR-T therapy (tisagenlecleucel) can be used as a substitute for intensive re-induction chemotherapy and HCT in patients with a germline CHEK2 mutation.
Checkpoint kinase 2 (CHEK2) is a tumor suppressor gene that plays a crucial role in the cell cycle and response to DNA damage induced by replication stress and double stranded DNA breaks. CHK2 kinase, a protein coded by the CHEK2 gene, is also required during mitosis for spindle formation, accurate attachment of kinetochores, and subsequent proper chromosome segregation. Therefore, CHEK2 mutations can lead to aberrant proteins, contributing to errors in chromosome segregation and a higher frequency of unbalanced structural rearrangements (1). Whether the CHEK2 gene is a true cancer predisposition syndrome gene by itself remains a topic of debate, but emerging evidence suggests an important role in cancer susceptibility, especially in relation to breast cancers (2, 3). Truncating CHEK2 gene mutations are well-known pathogenic variants, but the clinical significance of missense variants are subject of ongoing research and their interpretation remains challenging (4). Heterozygous pathogenic germline mutations in CHEK2 (c.1100delC) have been associated with an increased risk of hereditary breast, prostate, kidney, thyroid, and colon cancers (5–7). Congenital CHEK2 inactivation is also associated with an increased risk of hematologic malignancies, mainly myeloid neoplasms like myelodysplastic syndrome (MDS) (8). A recent study showed a two times higher frequency of unbalanced structural chromosomal rearrangements (58%) among patients harboring germline CHEK2 mutations, in comparison to non-carriers (27%) in MDS patients (9). However, lymphoid malignancies, especially B-ALL, seems to be a very rare occurrence in association with CHEK2 mutations. To the best of our knowledge, there has been no report on the use of tisagenlecleucel for relapsed B-ALL in a patient with a CHEK2 mutation (c.1100delC). Herein, we report the successful use of tisagenlecleucel for the treatment of early relapsed (both medullary and extramedullary) pediatric B-ALL in a patient with a CHEK2 mutation and papillary thyroid carcinoma.
A 12-year-old boy with a history of hypothyroidism presented with generalized petechiae and hematuria. The patient's white blood count at presentation was 446,750/microliter with mild anemia (hemoglobin 10.6 g/dl), severe thrombocytopenia (24,000/microliter) and 90% B-lymphoblasts in peripheral blood. Lumbar puncture (LP) revealed no blasts in cerebrospinal fluid (CSF). Cytogenetic analysis of bone marrow (BM) blasts revealed a normal karyotype, but fluorescent in situ hybridization (FISH) analysis revealed a 93% positivity for CRLF2::P2RY8 rearrangement. The patient subsequently started therapy as per Children's Oncology Group AALL1131 chemotherapeutic regimen. Family history revealed a diagnosis of metastatic thyroid cancer in a maternal uncle at the age of 44 years who died at the age of 45 years (Figure 1).
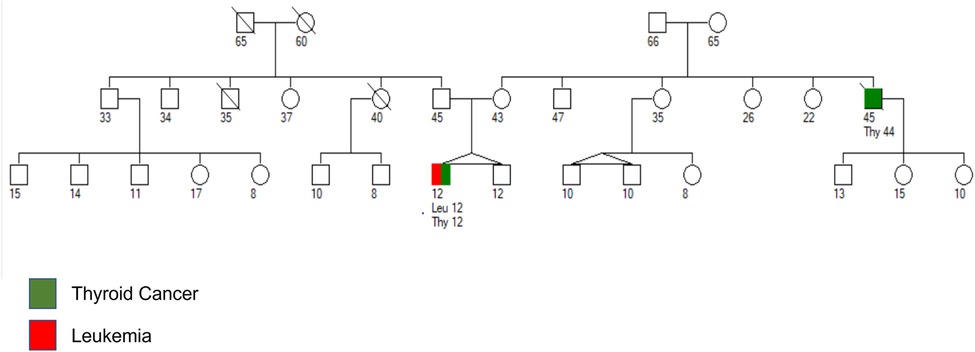
Figure 1. Pedigree chart with age of the patient and family members in years. Maternal uncle was diagnosed with anaplastic thyroid cancer at the age of 44 years and died from metastatic disease at the age of 45 years. Patient was diagnosed with papillary thyroid carcinoma and B-ALL at the age of 12 years.
Seven months after the diagnosis of B-ALL, he presented with a thyroid mass. Fine needle aspiration cytology was diagnostic of papillary thyroid cancer, staged at Bethesda Category V (Figure 2A). Evaluation for metastatic disease was negative. The patient underwent a total thyroidectomy with central lymph node biopsy followed by radioactive iodine therapy. He was referred to Genetics due to concerns for a cancer predisposition syndrome given the development of multiple malignancies within a year. Genetic testing revealed a pathogenic heterozygous CHEK2 gene mutation (c.1100delC; p.Thr367Metfs*15). The genetic panel also identified three variants of unclear significance: CDKN2A (c.37 °C > T;p. Arg124Cys), FLCN (c.62G > A; p.Cys21Tyr) and SDHAF2 (c.139A > G; p.Met47Val). A whole-body MRI was negative for any other malignancies.
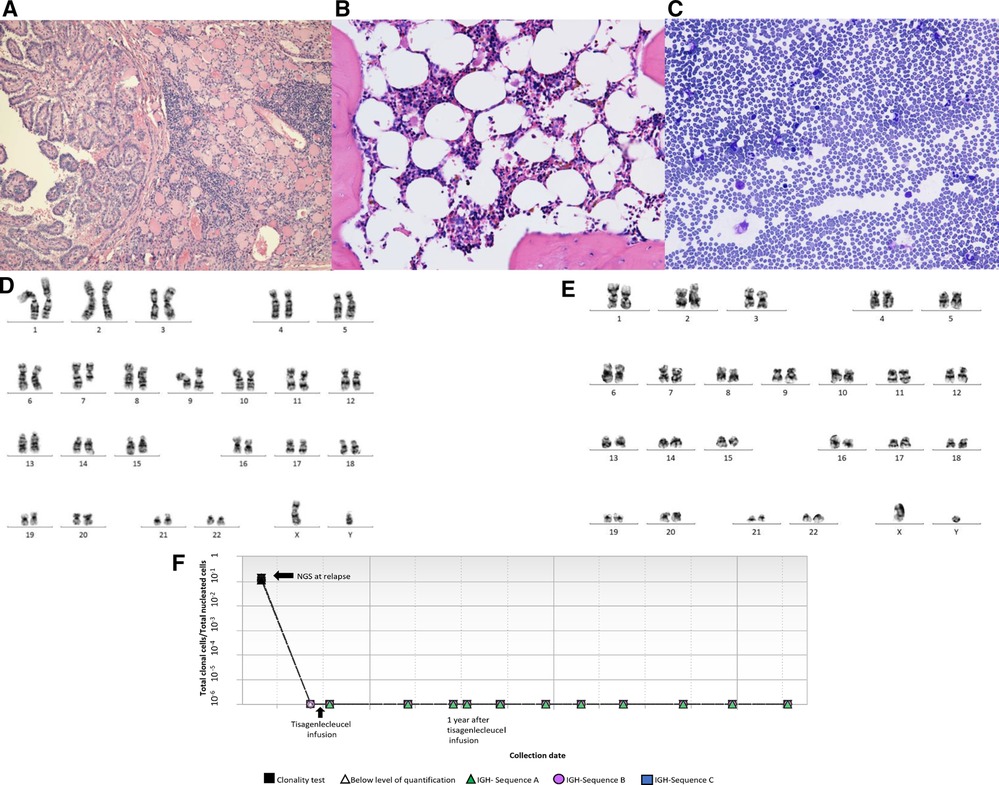
Figure 2. (A) Thyroidectomy reveals thyroid tissue with focal chronic inflammation and a papillary carcinoma (H&E stain, 100×). (B) Representative bone marrow biopsy reveals hypocellular marrow with trilineage hematopoiesis and no overt dysplasia (H&E stain, 400×). (C) Representative bone marrow aspirate reveals few erythroid and granulocytic cells with no overt dysplasia (Giemsa stain, 400×). (D) Karyotyping showing deletion of 7q. (E) Karyotyping showing absence of deletion 7q in most recent bone marrow specimen. (F Next generation sequencing (NGS) showing detection of leukemic clones at relapse and remission (0 residual clonal cells) at 1, 2 and 3 years after receiving tisagenlecleucel.
Fifteen months after his initial diagnosis, while on maintenance chemotherapy, the patient developed a frontotemporal headache. Computed tomography (CT) scan of the brain showed no intracranial involvement. Mild swelling of optic nerve head was noted on ophthalmologic examination. Increased intracranial pressure and central nervous system (CNS) disease was confirmed by lumbar puncture and magnetic resonance imaging of the brain. Cerebrospinal fluid studies revealed elevated WBC (48/microliter) with 75% lymphoid blasts. Bone marrow aspirate revealed 15% lymphoid blasts by flow cytometry with FISH positive for CRLF2::P2RY8 rearrangement suggesting relapse with the same leukemic clone that was found on initial diagnosis. Both BM and CSF lymphoid blasts were positive for CD19. He had no signs of any other extramedullary site involvement. He was started on bi-weekly triple intrathecal chemotherapy, in addition to systemic salvage chemotherapy consisting of vincristine, dexamethasone, and mitoxantrone for the next several weeks. The patient was eventually negative for CNS disease sixteen months after initial diagnosis.
In preparation for CAR-T therapy the patient underwent apheresis. The patient was then initiated on a bridging therapy with non-escalating Capizzi protocol prior to receiving tisagenlecleucel. Pre-CAR-T infusion BM and CSF evaluation (done prior to starting lymphodepletion) showed no blasts by flow cytometry. He subsequently received lymphocyte-depleting chemotherapy with fludarabine and cyclophosphamide prior to his infusion of tisagenlecleucel. Four days after CAR-T infusion, the patient developed grade 2 cytokine release syndrome (CRS) but did not develop any immune-effector cell associated neurotoxicity syndrome as per American Society for Transplantation and Cell Therapy Consensus Grading. The patient received tocilizumab and a short course anakinra (3 days). A CSF study was done on day 5 for severe headache in the setting of high-grade fever not responding to tocilizumab, which revealed 70 WBCs/uL with 72% lymphocytes (7% CD19 positive lymphoblasts along with atypical lymphocytes). Day 30 BM evaluation was negative for minimal residual disease by flow cytometry and confirmed by next generation sequencing (NGS) by clonoSEQ (Adaptive). CSF studies at the time showed increased lymphocytes but no blasts. Follow up LPs and BM aspirate/biopsies since then have been consistently negative for lymphoid blasts by flow cytometry. Of note, chromosomal analysis by karyotyping of the patient's bone marrow aspirate, taken 15 months post CAR-T, showed two abnormal clones in metaphase cells. The first clone revealed that 17% of cells had an unbalanced rearrangement of chromosome 7q, which resulted in a partial deletion of 7q, an anomaly frequently associated with primary and secondary myeloid disorders, including MDS. However, no morphologic evidence of myelodysplasia was noted on BM biopsy (Figures 2B,C). The second clone revealed that 13% of cells had a balanced reciprocal translocation (1;19) (q23;p13.3), which results in a TCF3-PBX1 fusion. However, concurrent interphase FISH studies were negative for this fusion. Serial BM biopsies and cytogenetic testing continues to reveal intermittent partial deletion of the distal portion of 7q (Figures 2D,E), while the findings of translocation (1;19)(q23;p13.3) have resolved. His B-ALL remains in remission with no abnormal clones identified on serial NGS testing (Figure 2F) or by immunophenotyping (flow cytometry) and has persistent B cell aplasia. Due to prior history of intensive chemotherapy, germline CHEK2 mutation and deletion 7q, we suspected a diagnosis of MDS but the follow up CBCs and BM examinations did not confirm our diagnosis due to following reasons. Firstly, the patient's complete blood count improved during the follow up and did not show any progression of cytopenia. Secondly, the chromosomal abnormality of deletion 7q was detected intermittently at a very low percentage that was only detected by karyotyping but not by FISH. Finally, this abnormality was not detected on the patient's most recent bone marrow examination (55 months after initial diagnosis of B-ALL), both on FISH and cytogenetics. We continue to follow this patient closely with serial (every 3- 6 month) CBCs and bone marrow examinations. A summary of the patient's clinical course can be found in Figure 3.
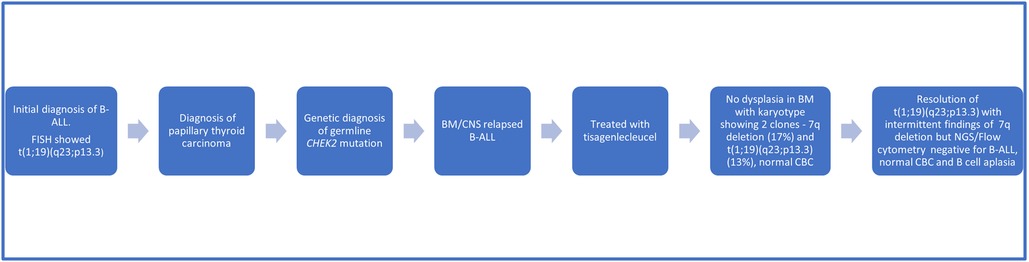
Figure 3. Timeline of clinical events.
Checkpoint kinase 2 is a tumor suppressor gene that plays a crucial role in the cell cycle and response to DNA damage through the ATM-CHEK2-p53 DNA damage response pathway (1). While truncating loss of function (LoF) mutations are known to be pathogenic, several variants of unknown significance have been described with undetermined functional and clinical significance. Homozygous LoF CHEK2 mutations can present as Li-Fraumeni (LFS)-like syndrome, whereas heterozygous LoF CHEK2 variants are moderate penetrance risk factors for solid tumors (2–7). Not surprisingly, emerging evidence also supports the association of certain CHEK2 gene mutations with myeloid and lymphoid malignancies. LoF CHEK2 mutations are now increasingly recognized as risk factors for myeloid malignancies (myeloproliferative neoplasms, myelodysplastic syndromes, and acute myeloid leukemia) (8, 9). Interestingly, pre-clinical murine models suggest a role of CHEK2 mutations (CHEK2 c.1100delC allele) in development of hematopoietic malignancies besides other solid tumors like breast and lung cancer (10). This is supported by clinical findings in large scale epidemiological studies of these malignant neoplasms (5–7). It must be noted that our patient had other cytogenetic abnormalities and molecular mutations besides CHEK2 mutation that most likely led to the development of B-ALL, and it is unlikely that the CHEK2 mutation was the primary driver mutation. However, given the evidence supporting association of CHEK2 mutation with MDS, the karyotypic findings of clones harboring MDS defining cytogenetic findings (partial deletion of the distal portion of 7q) is concerning for an increased risk of MDS in the future. Therefore, the patient is undergoing close observation with yearly bone marrow examination besides screening for other solid tumors.
The finding of germline CHEK2 mutation in a patient with relapsed/refractory hematological malignancy has profound clinical implications and presents several challenges. Choosing matched sibling donor (MSD) with the same germline CHEK2 mutation may present as a clinical dilemma, even though there is no definitive evidence to suggest relapse of leukemia or graft failure after MSD HCT. In our patient we avoided HCT due to availability of CAR-T therapy, the possible need for additional chemotherapy for CHEK2 mutation-associated solid tumors in the future, and the risk of therapy-related MDS or other secondary malignancies. The use of tisagenlecleucel has been previously described in a patient with LFS and B-ALL, but it has not been described in patients with CHEK2 mutations (11). Also, the patient did not have a well-matched unrelated donor or umbilical cord blood units. The patient's mother and fraternal twin brother also harbored the same CHEK2 gene mutation, though the father did not. However, father was not available as a potential donor. Therefore, haploidentical HCT was not considered a viable option. Some reports suggest that relapsed/refractory pediatric B-ALL treated with HCT after CAR-T infusion has significantly better median overall survival than the non-transplant group, especially if post CAR-T NGS is positive even at low levels (12, 13). However, the utility of HCT for patients in deep remission showing no (zero) leukemic clones remains uncertain and could probably be observed without consolidation with HCT (13). Therefore, in our patient with NGS negative remission, we have taken the approach of close monitoring with NGS testing without pursuing HCT. In the future, novel agents like poly (ADP-ribose) polymerase (PARP) inhibitors may be a potential therapeutic option for patients with ATM or CHEK2 mutation associated malignancies (8). The patient's fraternal twin and mother have also undergone genetic counseling and been educated regarding the need for cancer surveillance.
In summary, we describe the case of a pediatric patient with a heterozygous CHEK2 mutation (c.1100delC (p. Thr367Metfs*15) who presented with high-risk B-ALL and papillary thyroid cancer at the age of 12years. The patient's early relapsed B-ALL with bone marrow and CNS involvement was successfully treated with tisagenlecleucel. The patient responded and tolerated tisagenlecleucel very well. He continues to be in NGS-negative remission 3 years after treatment. The significance of intermittent findings of deletion of 7q on serial BM examinations in the absence of cytopenia and morphologic evidence of dysplasia remains a diagnostic dilemma and does not fulfill the criteria for diagnosis of MDS. However, it is concerning for potential development of either CHEK2 mutation-associated or therapy-related MDS in future. Hence, we continue to follow this patient closely with serial (every 3- 6 month) CBCs and bone marrow examinations. Moreover, the implication of infused CHEK2 mutations harbored by autologous CAR-T cells in the long term remains unknown. Our case highlights the need for more research in delineating the true role of pathogenic CHEK2 gene mutations in hematologic malignancies as a potential cancer predisposition gene. Finally, our case also illustrates that CAR-T therapy can be considered for treatment of R/R B-ALL patients with potential cancer predisposition syndromes such as CHEK2 mutation, in order to avoid HCT as has been previously described for a patient with LFS who developed B-ALL (11).
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by the study was exempt from ethical approval procedures. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
AI and AV: were involved in drafting, review, and revision of the initial and final manuscript. AA and DJ: were involved in the patient's treatment decision as well as the review and revision of the initial manuscript. JT and MB: were involved in genetic consultation as well as providing the pedigree chart, review, and revision of the initial manuscript. JC: performed pathologic examination of the bone marrow slides and provided pictures of bone marrow aspirate patient's karyotyping, next generation sequencing studies as well as the review and revision of the initial manuscript. All authors contributed to the article and approved the submitted version.
The authors thank the patient's family for providing consent and granting permission to publish this case report. We acknowledge the work of other medical staff of Children's National Hospital, Washington, DC who were associated with this patient in the preparation of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Stolarova L, Kleiblova P, Janatova M, Soukupova J, Zemankova P, Macurek L, et al. CHEK2 Germline variants in cancer predisposition: stalemate rather than checkmate. Cells. (2020) 9(12):2675. doi: 10.3390/cells9122675
2. Cybulski C, Górski B, Huzarski T, Masojć B, Mierzejewski M, Debniak T, et al. CHEK2 Is a multiorgan cancer susceptibility gene. Am J Hum Genet. (2004) 75(6):1131–5. doi: 10.1086/426403
3. Ruijs MW, Broeks A, Menko FH, Ausems MG, Wagner A, Oldenburg R, et al. The contribution of CHEK2 to the TP53-negative li-fraumeni phenotype. Hered Cancer Clin Pract. (2009) 7(1):4. doi: 10.1186/1897-4287-7-4
4. Boonen RACM, Wiegant WW, Celosse N, Vroling B, Heijl S, Kote-Jarai Z, et al. Functional analysis identifies damaging CHEK2 missense variants associated with increased cancer risk. Cancer Res. (2022) 82(4):615–31. doi: 10.1158/0008-5472.CAN-21-1845
5. Thompson D, Seal S, Schutte M, McGuffog L, Barfoot R, Renwick A, et al. A multicenter study of cancer incidence in CHEK2 1100delC mutation carriers. Cancer Epidemiol Biomarkers Prev. (2006) 15(12):2542–5. doi: 10.1158/1055-9965.EPI-06-0687
6. Huijts PE, Hollestelle A, Balliu B, Houwing-Duistermaat JJ, Meijers CM, Blom JC, et al. CHEK2*1100delC Homozygosity in The Netherlands–prevalence and risk of breast and lung cancer. Eur J Hum Genet. (2014) 22(1):46–51. doi: 10.1038/ejhg.2013.85
7. Koen K, Robin P, Eline N. CHEK2 Mutations and papillary thyroid cancer: correlation or coincidence? Hered Cancer Clin Pract. (2022) 20(1):5. doi: 10.1186/s13053-022-00211-7
8. Stubbins RJ, Korotev S, Godley LA. Germline CHEK2 and ATM variants in myeloid and other hematopoietic malignancies. Curr Hematol Malig Rep. (2022) 17(4):94–104. doi: 10.1007/s11899-022-00663-7
9. Janiszewska H, Bąk A, Skonieczka K, Jaśkowiec A, Kiełbiński M, Jachalska A, et al. Constitutional mutations of the CHEK2 gene are a risk factor for MDS, but not for de novo AML. Leuk Res. (2018) 70:74–8. doi: 10.1016/j.leukres.2018.05.013
10. Bahassi el M, Robbins SB, Yin M, Boivin GP, Kuiper R, van Steeg H, et al. Mice with the CHEK2*1100delC SNP are predisposed to cancer with a strong gender bias. Proc Natl Acad Sci U S A. (2009) 106(40):17111–6. doi: 10.1073/pnas.0909237106
11. Chen L, Xu B, Long X, Gu J, Lou Y, Wang D, et al. CAR T-cell therapy for a relapsed/refractory acute B-cell lymphoblastic lymphoma patient in the context of li-fraumeni syndrome. J Immunother Cancer. (2020) 8(1):e000364. doi: 10.1136/jitc-2019-000364
12. Summers C, Wu QV, Annesley C, Bleakley M, Dahlberg A, Narayanaswamy P, et al. Hematopoietic cell transplantation after CD19 chimeric antigen receptor T cell-induced acute lymphoblastic lymphoma remission confers a leukemia-free survival advantage. Transplant Cell Ther. (2022) 28(1):21–9. doi: 10.1016/j.jtct.2021.10.003
13. Pulsipher MA, Han X, Maude SL, Laetsch TW, Qayed M, Rives S, et al. Next-generation sequencing of minimal residual disease for predicting relapse after tisagenlecleucel in children and young adults with acute lymphoblastic leukemia. Blood Cancer Discov. (2022) 3(1):66–81. doi: 10.1158/2643-3230.BCD-21-0095
Keywords: CAR-T, tisagenlecleucel, B-ALL, CHEK2 mutation, MDS
Citation: Ipe A, Angiolillo A, Jacobsohn D, Cheng J, Bornhorst M, Turner J and Vatsayan A (2023) Case report: Tisagenlecleucel for treatment of relapsed B- acute lymphoblastic leukemia in a patient with CHEK2 mutation. Front. Pediatr. 11:1067131. doi: 10.3389/fped.2023.1067131
Received: 11 October 2022; Accepted: 25 January 2023;
Published: 1 March 2023.
Edited by:
Hasan Hashem, King Hussein Cancer Center, JordanReviewed by:
Kristian Schafernak, Phoenix Children's Hospital, United States© 2023 Ipe, Angiolillo, Jacobsohn, Cheng, Bornhorst, Turner and Vatsayan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abraham Ipe YWlwZUBnd3UuZWR1
Specialty Section: This article was submitted to Pediatric Hematology and Hematological Malignancies, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.