 Yang Yang
Yang Yang Zhu Wang
Zhu Wang Jia Chen
Jia Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 03 March 2023
Sec. Genetics of Common and Rare Diseases
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1059658
Rubinstein-Taybi syndrome (RSTS) is a rare autosomal dominantly inherited disease characterized by slow mental and physical growth, skeletal abnormalities (broad thumbs and big toes), and dysmorphic facial features. RSTS is associated with de novo variants of the epigenetic-associated gene CREBBP. RSTS is primarily diagnosed based on clinical manifestations and genetic testing. Cutis marmorata telangiectatica congenita (CMTC) is a rare, congenital, and typically benign vascular anomaly of unknown etiology; it is described as persistent reticulated marbled erythema. The diagnosis of CMTC is largely based on clinical features, and GNA11 mutations are associated with CMTC. In this case report, we describe the case of a preterm infant (boy) with RSTS and CMTC who had a novel frameshift mutation leading to a premature stop codon in the CREBBP gene. This study adds the novel mutation c.5837dupC to the known molecular spectrum of disease-causing CREBBP gene mutations.
Rubinstein-Taybi syndrome (RSTS), a rare autosomal dominantly inherited disease, is characterized by slow mental and physical growth, skeletal abnormalities (broad thumbs and big toes), and dysmorphic facial features (1). Other manifestations of RSTS include heart defects, genitourinary and central nervous system malformations, skin anomalies, and an increased predisposition to cancer (2). RSTS is mainly caused by de novo variants in the epigenetic-associated gene CREBBP (3). Moreover, the diagnosis of RSTS mainly depends on clinical manifestations and genetic testing.
Cutis marmorata telangiectatica congenita (CMTC) is a rare, congenital, and benign vascular anomaly of unknown etiology (4). It is described as a persistent reticulated, marbled erythema that blanches with pressure and does not resolve with heating. As it affects capillaries and venules, it is characterized by a slow-flow vascular lesion (5). The diagnosis of CMTC is primarily based on clinical features. Recently, GNA11 mutations have been reported to be associated with CMTC (6–8).
In the present study, we report the case of a preterm infant boy with RSTS and CMTC who presented with a novel frameshift mutation leading to a premature stop codon in the CREBBP gene. The child, with a typical facial appearance, experienced feeding difficulties, growth regression, and recurrent respiratory infections.
A boy, born at 34 weeks gestation, was transferred to our neonatal department after birth, requiring surfactant therapy and mechanical ventilation because of respiratory distress syndrome. The Apgar scores were 9–10–10. He weighed 2230 g (25th–50th percentile), was 43 cm long (10th–25th percentile), and his head circumference was 30 cm (10th–25th percentile). The patient presented with respiratory distress and polydactyly in both legs. In addition to the scalp, palm, sole, and mucous membranes, his entire body was covered with reticular marble-resembling flower-like macules (Figure 1). The skin changes did not disappear as the temperature in the environment increased. There was no family history of vascular malformation. No other congenital or neurological anomalies were observed. Repeat ultrasound examinations of the head and ophthalmological examinations were normal. Electroencephalography (EEG) and brain magnetic resonance imaging (MRI) revealed no abnormalities. Ear emission and auditory evoked potentials were normal, and echocardiography findings showed patent foramen ovale (PFO). Urinary ultrasound was normal, and thyroid function test results were all within normal limits. Due to respiratory distress and feeding difficulties, the patient stayed in the intensive care unit for approximately three months. Despite receiving antibiotics, respiratory support, and nasal feeding, his weight was 3800 g (<3rd percentile), his height was 53 cm (3rd–10th percentile), and his head circumference was 35 cm (<3rd percentile). In addition to microcephaly, he had arched eyebrows, down-slanting palpebral fissures, a broad nasal bridge, a triangular mouth, and broad big toes (Figure 1). At the age of four months, his Geisel score was equivalent to 4–8 weeks. In addition, the reticular marble-resembling flowers-like macules that covered his body didn't disappear.
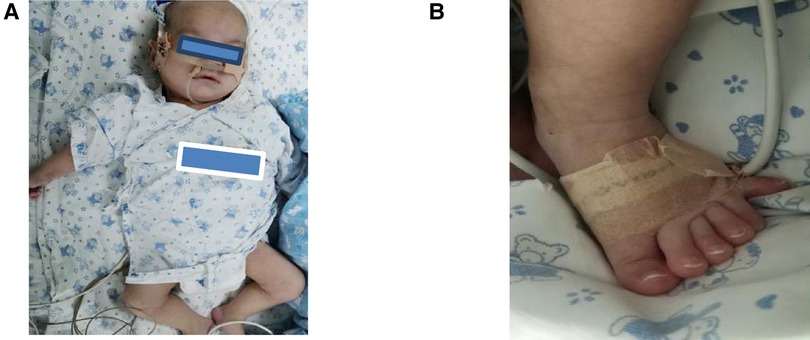
Figure 1. (A) A reticular erythematous path across the body, arched eyebrows, down-slanting palpebral fissures, a broad nasal bridge, and a triangular mouth. (B) The patient had polydactyly and broad big toes.
During the most recent follow-up, at the time of writing this manuscript, the patient was two years old, could only speak a few words, and could not walk independently.
The baby covered with reticulated marbled erythema had dysmorphic facial features, broad toes, and mental and developmental delays. Our hypothesis is a diagnosis of RSTS with CMTC. However, genetic testing is required to confirm.
When RSTS was suspected, the clinical findings were supplemented with CREBBP sequencing using next-generation genotyping techniques. Sequencing reactions were performed using the MiSeq Illumina sequencer, and data analysis was performed using the MiSeq Reporter. CREBBP sequencing revealed a C duplication at position c.5837 in the CREBBP gene (c.5837dupC), a frameshift mutation predicted to result in premature termination at the 1966th amino acid of the CREB-binding protein (p.Pro1947Thrfs*19). Neither parent had any abnormalities at this site (Figure 2). Therefore, RSTS was confirmed.
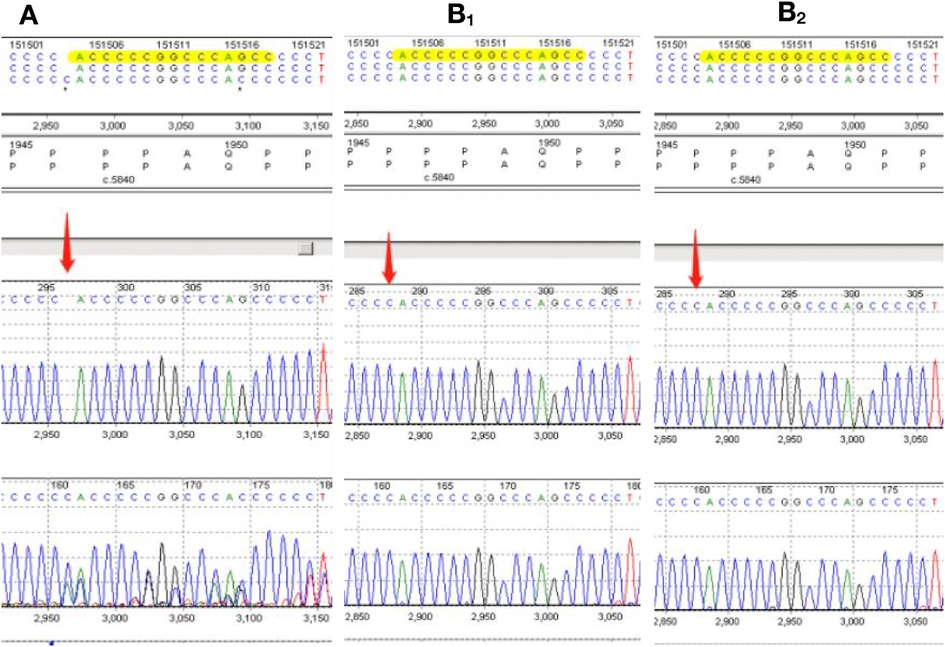
Figure 2. (A) sequence analysis of the genomic DNA of our rubinstein-taybi patient. Direct analysis of the CREBBP sequence reveals the C.5837dupC(p.P1974Thrfs*19) mutation. The CREBBP sequences of the patient's parents are also displayed (B1, B2).
RSTS, first described in 1963 by Rubinstein and Taybi (9), is caused by CREBBP mutations (10). To date, more than 500 CREBBP gene variants have been described in The Human Gene Mutation Database (HGMD) (3, 11). CREB-binding protein (CBP), encoded by CREBBP, is a histone and acetyl/lysine-transcription co-activator with more than 400 described interaction partners that regulate the expression of numerous genes during development and postnatal life (12). In this study, we present the case of an infant who presented with microcephaly, arched eyebrows, down-slanting palpebral fissures, a broad nasal bridge, a triangular mouth, broad big toes, polydactyly, postnatal developmental delay, and cognitive impairment.
Menke-Hennekam syndrome is characterized by the same facial dysmorphia as RSTS and is accompanied by epilepsy and hearing impairment. In our case, the baby had a big hallux and normal EEG and hearing tests. Moreover, Treacher-Collins syndrome, which is associated with mutations in the TCOF1, POLRIC, and POLRID genes, is marked by microcephaly, an intellectual deficit, and other congenital malformations (13). The main distinguishing features of the three diseases are a grimacing smile and broad toes or fingers (14,15). Large toes or fingers are also a manifestation of Pfeiffer syndrome, which is related to the FGFR1 or FGFR2 genes and neural structural malformations (13, 16). Moreover, Comelia de Lange and Floating-Harbour syndromes are also defined by intellectual deficits and other anomalies similar to RSTS; however, both are characterized by skeletal dysplasias such as those of the ulna and skull (13). In the present case, the brain MRI was normal, further confirming RSTS with CMTC.
RSTS can be accompanied by skin anomalies, with seven cases reported to date (11, 17). However, all of these reports dealt with neural crest-derived tumors, keloid or hypertrophic scars, supernumerary nipples, ingrown toenails, paronychia, hypertrichosis, and glabellar hemangiomas (11, 17). The c.5837dupC (p.Pro1947Thrfs*19) mutation in CREBBP identified in this study has also been detected in both a five-year-old girl and a 12-year-old girl with RSTS, and one of these girls developed pilomatricomas. In our case, RSTS was accompanied by CMTC. Although all three cases (ours and the two girls') had the same mutation, the phenotypes differed. Therefore, the same mutation may contribute to the disease via multiple molecular pathways. Furthermore, some studies have suggested a link between CMTC and mutations in GNA11 (5). However, in our case, CMTC was caused by a mutation in CREBBP.
The patient in our case study had polydactyly, similar to a previous report, and we discovered that these symptoms are extremely common in Chinese patients. However, this high incidence has rarely been reported in previous patients with RSTS (3). Moreover, studies addressing genotype-phenotype correlations do not detect statistically significant differences between RSTS cases with distinct mutations or between RSTS cases with and without mutations (18). However, our case report indicates that the mutations in the same gene may have different clinical manifestations in RSTS, warranting further investigation.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by the Ethics Committee and the institutional review board of the Guangdong Women and Children Hospital (Guangzhou, China). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
All Six authors are involved in the clinical care of the patient, and in the write up and editing of the submitted case report. JC searched for CMTC disease related information through children's skin texture features. YYY collected patient related photos and follow-up information, which provided information resources for case reports. JX contacted his family members and asked for advice on case reports. JWX and ZW persuaded his family members to complete the full exon examination. YY completed the writing of the whole case report. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Al-Qattan MM, Rahbeeni ZA, Al-Hassnan ZN, Jarman A, Rafique A, Mahabbat N, et al. Chromosome 16p13.3 contiguous gene deletion syndrome including the SLX4, DNASE1, TRAP1, and CREBBP genes presenting as a relatively mild rubinstein-taybi syndrome phenotype: a case report of a Saudi boy. Case Rep Genet. (2020) 2020:6143050. doi: 10.1155/2020/6143050
2. Eser M, Ayaz A, Yeşil G. A case with rubinstein-taybi syndrome: a novel frameshift mutation in the CREBBP gene. Turk J Pediatr. (2017) 59(5):601–3. doi: 10.24953/turkjped.2017.05.017
3. Yu S, Wu B, Qian Y, Zhang P, Lu Y, Dong X, et al. Clinical exome sequencing identifies novel CREBBP variants in 18 Chinese rubinstein-taybi syndrome kids with high frequency of polydactyly. Mol Genet Genomic Med. (2019) 7(12):e1009. doi: 10.1002/mgg3.1009
4. International Society for the Study of Vascular Anomalies. ISSVA Classification of Vascular Anomalies (2018). https.issva.org/classification [Accessed 28 May 2019].
5. Bui TNPT, Corap A, Bygum A. Cutis marmorata telangiectatica congenital: a literature review. Orphanet J Rare Dis. (2019) 14(1):283. doi: 10.1186/s13023-019-1229-8
6. Sassalos TM, Fields TS, Levine R, Gao H. Retinal neovascularization from a patient with cuit marmorata telangiectatica congenita. Retin Cases Brief Rep. (2018) 15(1):77–80. doi: 10.1097/ICB.0000000000000736
7. Thomas FP, Guergueltcheva V, Gondim FA, Tournev I, Rao CV, Ishpekova B, et al. Clinical, neurophysiological and morphological study of dominant intermediate charcot-marie-tooth type C neuropathy. J Neurol. (2016) 263(3):467–76. doi: 10.1007/s00415-015-7989-8
8. Kumar A, Zastrow DB, Kravets EJ, Beleford D, Ruzhnikov MRZ, Grove ME, et al. Extracutaneous manifestations in phacomatosis cesioflammea and cesiomarmorata: case series and literature review. Am J Med Genet A. (2019) 179(6):966–77. doi: 10.1002/ajmg.a.61134
9. Rubinstein JH, Taybi H. Broad thumbs and toes and facial abnormalities. A possible mental retardation syndrome. Am J Dis Child. (1963) 105:588–608. doi: 10.1001/archpedi.1963.02080040590010
10. Perij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RCM, Masuno M, et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. (1995) 376:348–51. doi: 10.1038/376348a0
11. Zhang B, Gong C. New progress in diagnosis and treatment of rubinstein taybi syndrome. Chin J Pediatr. (2021) 36(22):1746–50.
12. Spena S, Milani D, Rusconi D, Negri G, Colapietro P, Elcioglu N, et al. Insights into genotype-phenotype correlations from CREBBP point mutation screening in a cohort of 46 rubinstein-taybi syndrome patients. Clin Genet. (2015) 88(5):431–40. doi: 10.1111/cge.12537
13. Van Gils J, Magdinier F, Fergelot P, Lacombe D. Rubinstein-Taybi syndrome: a model of epigenetic disorder. Genes (Basel). (2021) 12(7):968. doi: 10.3390/genes12070968
14. Menke LA, van Belzen MJ, Alders M, Cristofoli F, Study DDD, Ehmke N, et al. CREBBP Mutations in individuals without rubinstein-taybi syndrome phenotype. Am J Med Genet A. (2016) 170(10):2681–93. doi: 10.1002/ajmg.a.37800
15. Luo GJ, Hou M, Yuan AY, Liu QY, Chen J. [A case of menke-hennekam syndrome-1 caused by CREBBP gene variation]. Zhonghua Er Ke Za Zhi. (2022) 60(10):1074–6. Chinese. doi: 10.3760/cma.j.cn112140-20220406-00291
16. Mavridis IN, Rodrigues D. Nervous system involvement in pfeiffer syndrome. Childs Nerv Syst. (2021) 37(2):367–74. doi: 10.1007/s00381-020-04934-7
17. Jager MJ, Shields CL, Cebulla CM, Abdel-Rahman MH, Grossniklaus HE, Stern M-H, et al. Uveal melanoma. Nat Rev Dis Primers. (2020) 6(1):24. doi: 10.1038/s41572-020-0158-0
Keywords: rubinstein-taybi syndrome, cutis marmorata telangiectatica congenita, CREBBP, case report, preterm
Citation: Yang Y, Xiao J, Ye Y, Xiang J, Wang Z and Chen J (2023) Case report: A preterm infant with rubinstein-taybi syndrome and Marmorata telangiectatica harboring a frameshift mutation in the CREBBP gene. Front. Pediatr. 11:1059658. doi: 10.3389/fped.2023.1059658
Received: 1 October 2022; Accepted: 16 January 2023;
Published: 3 March 2023.
Edited by:
Corrado Romano, University of Catania, ItalyReviewed by:
Elena Dominguez, Centro de Investigación Biomédica de La Rioja, Spain© 2023 Yang, Xiao, Ye, Xiang, Wang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia Chen NzQ1NzUwOTY0QHFxLmNvbQ==
Specialty Section: This article was submitted to Genetics of Common and Rare Diseases, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.