 Jianfang Zhu
Jianfang Zhu Yuxiao Sun
Yuxiao Sun Weiyan Zheng
Weiyan Zheng Chunlin Wang
Chunlin Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 30 March 2023
Sec. Children and Health
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1057574
This article is part of the Research Topic Precision Medicine of Non-Communicable Diseases in Children View all 10 articles
Gaucher disease (GD) is an inherited lysosomal storage disease caused by mutations in the glucocerebrosidase gene. The decrease of glucocerebrosidase activity in lysosomes results in the accumulation of its substrate glucocerebroside in the lysosomes of macrophages in organs such as the liver, spleen, bones, lungs, brain and eyes, and the formation of typical storage cells, namely “Gaucher cells”, leading to lesions in the affected tissues and organs. Hepatosplenomegaly, bone pain, cytopenia, neurological symptoms, and other systemic manifestations are common in clinical practice. Most pediatric patients have severe symptoms. Early diagnosis and treatment are crucial to improve the curative effect and prognosis. However, due to the low incidence of this disease, multi-system involvement in patients, and diverse clinical manifestations, multidisciplinary teamwork is needed for comprehensive evaluation, diagnosis and treatment. In this study, we reported 2 cases of different types of GD who were diagnosed, treated and followed up by multidisciplinary collaboration in infancy.
GD is a clinically rare single-gene autosomal recessive metabolic disorder. Due to the mutation of the glucocerebrosidase (GBA) gene, the activity of GBA in the lysosomes is reduced, which causes its substrate glucocerebroside to be stored in the lysosomes of macrophages in organs such as the liver, spleen, bones, lungs, brain and eyes to form “Gaucher cells”. The clinical manifestations are usually hepatosplenomegaly, bone pain, anemia, thrombocytopenia, neurological symptoms, and other systemic manifestations, which may progressively worsen during the course of the disease. There are three types of GD according to whether the nervous system is involved and the rate of disease progression (1–4). (1) Type I has historically been characterized by the absence of neurological involvement. Nevertheles, the prevalence of peripheral neuropathy in adults with type I has been reported to be higher than that in the general population (5). Evidence suggests that central nervous system (CNS) involvement may also occur in patients with type I (6, 7). Two third of type I develop the disease in childhood, and the severity of symptoms varies greatly. (2) Type II is an acute neuropathic type with extensive and severe visceral involvement, which usually develops within the first year after birth, and most patients die at the age of 2 to 4 years. (3) Type III is a subacute or chronic neuropathic type, which often develops in childhood. The early manifestations are similar to those of type I. The neurological symptoms of varying degrees of severity gradually appear with slow disease progression. In addition, there are rare subtypes such as perinatal lethal type and cardiovascular type (8).
The clinical symptoms and signs of GD are not characteristic, which can be insidiously in children or adults at different ages. Many medical diseases, such as leukemia, lymphoma, Niemanpick disease, and Langerhans cell histiocytosis, have similar manifestations to GD, which can easily lead to delayed diagnosis. A survey data from multiple countries (9) showed that many patients with GD experienced delays in diagnosis and misdiagnosis, and nearly 1/6 of the patients took more than 7 years to be diagnosed. Nevertheless, the two cases in this study were both diagnosed within six months of the onset of clinical symptoms. It is believed that with the continuous improvement of the understanding of rare diseases and the continuous development of diagnostic techniques, rare diseases including GD will be diagnosed and treated timely and accurately (10). In this study, we reported 2 cases of different types of GD who were diagnosed, evaluated, treated and followed up by multidisciplinary collaboration in infancy.
Hepatosplenomegaly, especially splenomegaly, is the main manifestation of digestive system involvement in GD, which is often accompanied by hypersplenism, sometimes giant spleen, splenic infarction, and splenic rupture (11). Hepatosplenomegaly may be asymptomatic or manifest as anorexia, abdominal distension, abdominal discomfort, or dull epigastric pain, and rarely, acute abdominal pain due to splenic infarction. In this study, both children firstly visited the department of gastroenterology because of abdominal distension. Case 1 was a female child, aged 1 year and 7 months (date of birth 2018.9, G1P1, full-term natural delivery, birth weight 3.2 kg, no history of asphyxia rescue). She went to the doctor because of “abdominal distension and appetite reduction for 3 months”. Physical examination findings were as follows: abdominal distention, prominent on the left side; tough and sharp edged liver 2 cm below the ribs; splenomegaly, 3 cm below the umbilicus, with the lower margin close to the groin, hard in texture, no obvious swelling on the surface; normal liver function and coagulation function, negative for hepatotropic virus, normal serum copper, ceruloplasmin, ferritin, transferrin saturation and total iron binding capacity. Abdominal ultrasound showed that the right hepatic lobe was 2.2 cm below the ribs with the oblique diameter of 8.3 cm, and there were a smooth and complete capsule, uniform intrahepatic echoes, no obvious abnormal echoes, no obvious dilation of the intrahepatic bile duct, and clear intrahepatic vascular orientation. The spleen was half-moon-shaped and evenly hypoechoic, with a smooth and complete capsule, intercostal thickness of 4.6 cm, the lower margin 3.5 cm below the umbilicus, and 3.7 cm across the midline at the right margin. The gallbladder and pancreas were normal. Enhanced CT of the whole abdomen also indicated hepatosplenomegaly. Case 2 was a male child, aged 1 year (date of birth 2019.4, G1P1, full-term cesarean section, birth weight 3.6 kg, no history of asphyxia rescue). He went to the doctor because of “abdominal distension and appetite reduction for 1 month”, with abdominal distention and obvious enlargement of liver and spleen. Auxiliary examination revealed normal liver function and coagulation function, negative for hepatotropic virus, and normal serum copper, ceruloplasmin, ferritin, transferrin saturation and total iron binding capacity. Abdominal ultrasound showed that the right hepatic lobe was 2.8 cm below the ribs with the oblique diameter of 7.8 cm, and the size of the left liver was about 5.0*6.6 cm. The spleen was large in shape with thickness of 3.2 cm, 5.4 cm below the ribs, with a smooth and complete outline, and the even and delicate echoes of spleen parenchyma. The gallbladder and pancreas were normal. Abdominal CT revealed hepatomegaly, splenomegaly and enlarged hilar lymph nodes. After treatment with enzyme replacement therapy (ERT), the hepatosplenomegaly of both children was significantly improved, and the liver function was within the normal range.
The main manifestation are cytopenia (anemia, thrombocytopenia, leukopenia, and agranulocytosis may occur alone or simultaneously) and bleeding tendency (increased bleeding tendency in GD patients is associated with thrombocytopenia, abnormal coagulation and platelet function defects). Two children in this study had mild anemia and thrombocytopenia (case 1: 96 g/L, 71 × 10E9/l; case 2: 101 g/L, 98 × 10E9/L), and the leukocytes and coagulation function were within the normal range. Anemia and thrombocytopenia gradually recovered within 6 months of ERT in both two patients. Bone marrow biopsy was performed in both children by the department of hematology, and Gaucher cells were found in both cases (Figure 1). The enzyme activity test showed that β-GBA in case 1 was 3.1 nmol/mg·h, and β-GBA in case 2 was 1.2 nmol/mg·h, both significantly lower than the normal reference range (6.56∼55.1 nmol/mg·h). GD genetic testing reported that case 1 was a homozygous mutation of GBA gene c.1448 T > C (p.L483P), and case 2 was a compound heterozygous mutation [c.475C > T (p.R159W) heterozygous mutation (from mother); c.1448 T > C (L483P) heterozygous mutation (from father)]. The above mutation sites are all known pathogenic mutations (12).
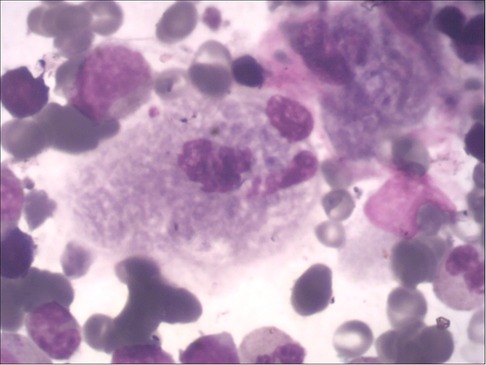
Figure 1. Gaucher cells in the bone marrow, with large or giant cell bodies, small nuclei, nuclear deformities, multinucleation, and dense chromatin. The cytoplasm is abundant and arranged in a gray-blue “onion skin” pattern.
GD can involve the whole-body bone with varying degrees of severity. The pathological change is the deposition of Gaucher cells in the bone, destroying and replacing the normal bone tissue. The affected parts mainly include the early lumbar spine, the metaphysis of long bones, the diaphysis, and the epiphysis in the middle and late stages. Patients with osteonecrosis often have acute/chronic bone pain, and may develop sudden local pain, swelling, or fever, and even aseptic osteomyelitis (13). In this study, both children had bone destruction. In case 1, bone imaging revealed low density lesions in the medial cortex of the right humerus and uneven bone density in both iliac bones. The remaining long bones of the limbs and spine were in normal shape, with continuous bone cortex and no abnormal changes in bone density. In case 2, the bone imaging examination showed that the left radius was unnatural, with local thickening of the cortical bone, and the x-rays of the remaining long bones of the limbs showed no obvious abnormality. The long bone destruction of the limbs occurred during the treatment and follow-up (Figure 2).
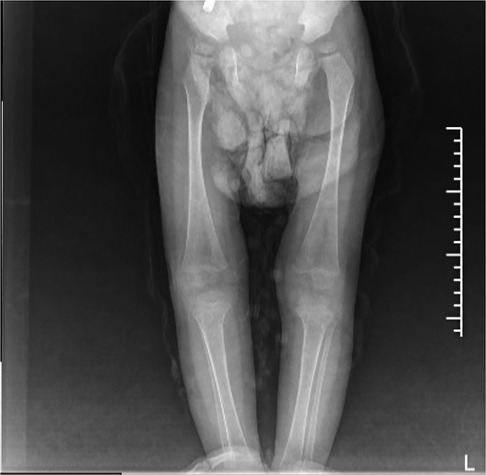
Figure 2. Signs of GD on both sides of the femur and tibia on October 21, 2021: enlarged and flask-shaped distal femur and proximal tibia on both sides, widened bone marrow cavity, and reduced bone density. No abnormal signs were observed in soft tissue.
Endocrine involvement in GD mainly involves abnormal growth and development, bone metabolism, nutritional status and glucose metabolism. GD patients with childhood/adolescent onset are prone to growth retardation and a higher risk of malnutrition (14). In this study, 2 children were evaluated by the department of endocrinology at the beginning of the disease. Case 1 was severely growth retardation and development and malnutrition [height 71 cm (-3.21SDS), weight 7.5 kg (-2.8SDS); patent anterior fontanelle, 0.5*0.5 CM, head circumference 44 cm, chest circumference 46 cm, subcutaneous fat 0.3 CM] with normal bone metabolism and glucose metabolism indexes. Case 2 has moderate growth retardation and malnutrition [height 70 cm (-2.41SDS), weight 8 kg (-1.95SDS); patent bregma, 1.0*1.0 CM, head circumference 43 cm, bust circumference 45 cm, subcutaneous fat 0.4 CM] with normal bone metabolism and glucose metabolism indexes. After the primary disease treatment, nutritional guidance was given on carbohydrates (carbohydrates with low glycemic index are preferred, sugary foods are avoided, and foods rich in dietary fiber are adequately consumed), protein (referring to the recommended intake of protein for the same age, based on high-quality proteins such as animal and soy proteins), fat (appropriate intake of fat, limited intake of saturated fatty acids, and increasing the intake ratio of monounsaturated fatty acids and polyunsaturated fatty acids), minerals and vitamins (supplementation of vitamin D and calcium according to monitoring indicators). At 2.3 years of follow-up, case 1 (3 years and 11 months) was evaluated as mild growth retardation [height 96 cm (−1.67SDS), weight 13 kg (−1.73SDS); head circumference 48 cm, chest circumference 52 cm, subcutaneous fat 0.5 cm] with normal bone metabolism and glucose metabolism indexes. Case 2 (3 years and 3 months) was evaluated as severely retarded in growth and development [height 82 cm (−4.33SDS), weight 9 kg (−3.91SDS); head circumference 45 cm, chest circumference 48 cm, subcutaneous fat 0.3CM].
Type I GD has no significant manifestation of primary central nervous system involvement in childhood, while type II and type III GD are neuropathic type associated with nervous system involvement. Type II is an acute neuropathic type, with onset from the neonatal period to infancy, mainly manifesting as early-onset and rapidly progressive neurological involvement, such as bilateral fixed strabismus, oculomotor nerve palsy, sucking and swallowing difficulties, as well as epilepsy, opisthotonos, and cognitive disorders. Patients with type II GD usually die at the age of 2 to 4 years (15). Type III is chronic or subacute neuropathy type, and the early manifestation is similar to type I. The neurological symptoms of varying degrees of severity gradually appear with slow disease progression, even remain mild throughout their lives. Patients may have oculomotor involvement, horizontal eye movement disorders and ataxia, epilepsy, myoclonic seizures, developmental delay, intellectual disability (15, 16). There are usually electroencephalogram abnormalities, most of which are characterized by slow wave background and interictal epileptiform discharges (sharp wave, spike wave, multiple spike wave, spike-slow complex wave, etc.) (17). In this study, both children were evaluated by the department of neurology. Case 1 had no positive signs in nervous system examination, no abnormality in head MRI and EEG, and no nervous system involvement, and was diagnosed as type I GD. Case 2 had onset in infancy with delayed language and motor development, and early neurological symptoms (initially shaking limbs during sleep, then rapidly progressing to tonic convulsions and frequent seizures). Initial evaluation of head MRI showed bilateral frontotemporal extracerebral space widening (Figure 3A), and EEG showed that the EEG activity was relatively depressed (low amplitude and lack of changes). Auditory brainstem evoked potentials suggested hearing impairment, later EEG showed epileptiform EEG changes, and head MRI showed brain atrophy (Figure 3B). Case 2 died due to uncontrolled status epilepticus and ineffective treatment at the age of 3 years and 3 months (2022.8.10), and was diagnosed as type II GD.
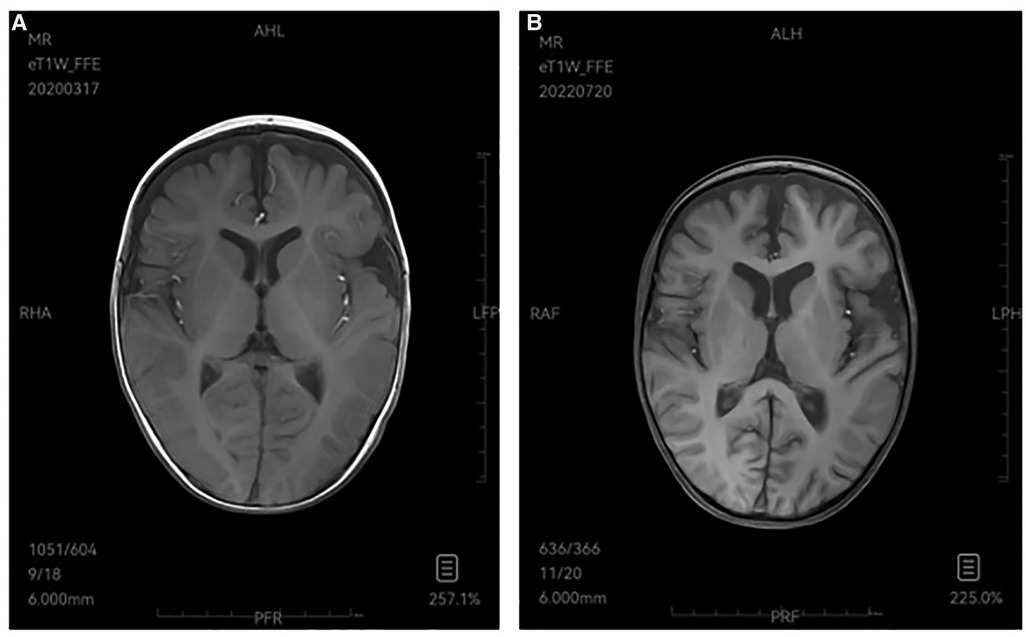
Figure 3. (A) 2020-3 MRI-T1W image demonstrates bilateral frontotemporal extracerebral space widening. (B) 2022-7 MRI-T1W image shows brain atrophy.
There are relatively few studies on ophthalmic involvement in GD. Some patients may have symptoms such as retinopathy, corneal opacity, uveitis, pinguecula and conjunctival thickening, abnormal eye movements, and strabismus (18, 19). No ophthalmic involvement was found in both children in this study by Slit-lamp microscopy, fundus photography, and other ophthalmic examinations.
Cardiac involvement in GD is extremely rare, and only case reports have been reported. It mainly occurs in subtype c of type III GD, which occours in the homozygous D409H genotype (20). Cardiomyopathy and valvular disease are the manifestations of involvement, and there are also structural and functional abnormalities of the right heart secondary to pulmonary hypertension, which can be clinically manifested as cardiac failure, arrhythmia, and increased incidence of thromboembolic events with atrial enlargement (21). In this study, both children were evaluated by ECG, cardiac ultrasound, and myocardial enzyme spectrum, and no cardiac involvement was observed.
Gaucher cells may directly infiltrate lung parenchyma and pulmonary vessels to cause pulmonary hypertension or interstitial lung disease. Pulmonary function examination mainly shows the decline of diffusion function, accompanied by varying degrees of restrictive ventilatory dysfunction. Imaging may show signs of pulmonary interlobular septal thickening, ground glass-like changes, reticular nodule infiltration, air retention and bronchiectasis (22, 23). Due to the obvious symptoms related to the involvement of extrapulmonary organs, respiratory symptoms are easily overlooked, which should be paid attention to by clinicians. GD patients with p.L483P gene mutation are prone to respiratory system involvement (24). In case 1, there were no abnormalities in the monitoring of oxygen saturation, determination of serum markers of pulmonary fibrosis, pulmonary function and pulmonary CT examination from initial diagnosis to follow-up. In case 2, pulmonary CT showed a small amount of inflammation in the upper lobes of both lungs at the first visit, pulmonary function test indicated mild mal-ventilation, and serum markers of pulmonary fibrosis showed elevated collagen IV. Pulmonary inflammation was aggravated several times during treatment and follow-up, which was improved by anti-infection and supportive treatment. It may suggest that ERT does not improve the occurred lung damage.
Different from other storage diseases, renal involvement in GD is rare, and clinical manifestations include proteinuria, microscopic hematuria, renal tubular dysfunction, and renal insufficiency or even failure (3). Both children in this study were evaluated by routine urinalysis and renal function tests, and no renal involvement was observed.
GD patients with ischemic splenic infarction due to enlarged spleen may develop acute abdominal pain, fever, and inflammation around the spleen (3). For those with giant spleen or hypersplenism, splenectomy can significantly improve clinical symptoms, reduce anemia and bleeding tendency, and improve developmental status (25). There was no obvious anemia and bleeding tendency caused by hypersplenism in both children in this study, and no surgical intervention was performed.
Specific treatments for GD mainly include ERT, hematopoietic stem cell transplantation (HSCT), substrate reduction therapy (SRT), molecular chaperone therapy and gene therapy. Among them, ERT can specifically supplement the enzyme lacking in the patient's body and reduce the storage of glucocerebroside in the body, which is the standard treatment for GD (26–28). The mechanism of action of SRT is to inhibit substrate formation and directly reduce substrate accumulation in cells. Currently, SRT is only applicable to adults, not children (29). Gene therapy, which is still in the clinical research phase and needs to be evaluated.
As the first-line treatment of GD, ERT can shrink the volume of the liver and spleen, improve anemia, thrombocytopenia, relieve bone pain, maintain normal growth and development, and improve quality of life (26–29). However, ERT drugs are macromolecules that cannot penetrate the blood-brain barrier and cannot improve neurological symptoms (15).
In this study, case 1 was diagnosed as type I GD at the beginning of the disease after multidisciplinary discussions. Due to high risk factors such as severe growth retardation and skeletal imaging changes (30), cerezyme was given intravenously at 60 U/kg, once every 2 weeks. The volume of the liver and spleen was significantly reduced during the 2.3-year treatment and follow-up. Anemia and thrombocytopenia gradually recovered within 6 months of treatment, and three types of hemocytes remained within the normal range. The indexes of physical development gradually improved from severe growth retardation to mild level, and the monitoring indicators of bone metabolism and glucose metabolism had always been within the normal range. No progressive damages to bones and no fractures occurred. The nervous system, cardiovascular system, respiratory system, renal system and eyes were not damaged. The quality of life improved significantly.
Case 2 could not be classified because of mild neurological involvement in the early stage. After multidisciplinary discussion, considering the existence of high-risk factors such as growth retardation, skeletal imaging changes, and neurological symptoms, cerezyme was given intravenously at 60 U/ kg, once every 2 weeks, and nutritional support was given at the same time. The volume of the liver and spleen was significantly reduced during the treatment and follow-up. Anemia and thrombocytopenia recovered gradually within 6 months of treatment. However, the indexes of physical development did not improved, and the growth retardation gradually progressed from moderate to severe level. There were also progressive damages to the bones, but no fractures occurred. The damages to the central nervous system progressed rapidly with frequent convulsions and brain atrophy. The patient died at the age of 3 years and 3 months after poor anti-epileptic treatment. There was mild mal-ventilation in the lungs at the beginning of the disease, and pulmonary inflammation was aggravated several times during the treatment and follow-up, and anti-infective treatment was given. The cardiovascular system, renal system and eyes were not damaged. According to the early involvement, rapid progression of the nervous system, and multiple organ damages, the patient was diagnosed as type II GD.
GD is a disease involving multiple systems, requiring multidisciplinary cooperation in the diagnosis and treatment process. In addition to the above clinical departments, more departments such as clinical laboratory, imaging department, genetic testing, pathology, pharmacy and nutritional department are needed to accurately diagnose and treat, improve the prognosis and quality of life, and reduce the risk of skeletal and other systemic disabilities (31). In addition, there is currently no effective treatment for children with type II GD.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by The Tab of Animal Experimental Ethical Inspection of the First Affiliated Hospital, Zhejiang University School of Medicine. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
JZ, YS, WZ and CW contributed to conception and design of the study. YS organized the database. JZ wrote the first draft of the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by Natural Science Foundation of Zhejiang Province (No. LQ19H310002).
The authors sincerely thank and acknowledge colleagues in clinical and auxiliary departments for their support on the diagnosis and treatment of the 2 children. And we also thank the children and parents for their persistence.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Subspecialty Group of Endocrinologic, Hereditary and Metabolic Diseases, the Society of Pediatrics, Chinese Medical Association; Subspecialty Group of Hematology, the Society of Pediatrics, Chinese Medical Association; Erythrocyte Disorders (Anemia) Group of Hematological Branch of Chinese Medical Association. China 2015 Expert consensus on Gaucher disease for diagnosis and treatment. Zhonghua Er Ke Za Zhi. (2015) 53:256–61. doi: 10.3760/cma.j.issn.0578-1310.2015.04.006
2. Subspecialty Group of Endocrinologic, Hereditary and Metabolic Diseases, the Society of Pediatrics, Chinese Medical Association; Subspecialty Group of Hematology, the Society of Pediatrics, Chinese Medical Association; Society of Medical Genetics, Chinese Medical Association; China Alliance for Rare Diseases. Expert consensus on diagnosis and treatment of pediatric Gaucher disease (2021). Zhonghua Er Ke Za Zhi. (2021) 12:1025–31. doi: 10.3760/cma.j.cn112140-20210611-00494
3. Erythrocyte Disorders (Anemia) Group of Hematological Branch of CMA. Expert consensus on the diagnosis and treatment of adult Gaucher disease in China. Chin J Med. (2020) 24:1841–9. doi: 10.3760/cma.j.cn112137-20200222-00405
4. Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, et al. A review of gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci. (2017) 18:441. doi: 10.3390/ijms18020441
5. Biegstraaten M, Mengel E, Marodi L, Petakov M, Niederau C, Giraldo P, et al. Peripheral neuropathy in adult type 1 Gaucher disease: a 2-year prospective observational study. Brain. (2010) 133:2909–19. doi: 10.1093/brain/awq198
6. Mullin S, Hughes D, Mehta A, Schapira AHV. Neurological effects of glucocerebrosidase gene mutations. Eur J Neurol. (2019) 26:388–93. doi: 10.1111/ene.13837
7. Bultron G, Kacena K, Pearson D, Boxer M, Yang R, Sathe S, et al. The risk of Parkinson's disease in type 1 Gaucher disease. J Inherit Metab Dis. (2010) 33:167–73. doi: 10.1007/s10545-010-9055-0
8. Puri RD, Kapoor S, Kishnani PS, Dalal A, Gupta N, Muranjan M, et al. Diagnosis and management of Gaucher disease in India-consensus guidelines of the Gaucher disease task force of the society for Indian academy of medical genetics and the Indian academy of pediatrics. Indian Pediatr. (2018) 2:143–53. doi: 10.1007/s13312-018-1249-9
9. Mehta A, Belmatoug N, Bembi B, Deegan P, Elstein D, Göker-Alpan Ö, et al. Exploring the patient journey to diagnosis of Gaucher disease from the perspective of 212 patients with Gaucher disease and 16 Gaucher expert physicians. Mol Genet Metab. (2017) 3:122–9. doi: 10.1016/j.ymgme.2017.08.002
10. Multi-disciplinary Team for Rare Diseases, Peking Union Medical College Hospital. Expert consensus on the multidisciplinary diagnosis and treatment of Gaucher disease. Med J Peking Union Med Coll Hosp. (2020) 6:682–97. doi: 10.3969/j.issn.1674_9081.2020.06.010
11. Zimran A. How I treat gaucher disease. Blood. (2011) 118:1463. doi: 10.1182/blood-2011-04-308890
12. Sun XY, Xue Y, Wang YP, Huang J, Lin RF, Kang MY, et al. Clinical phenotype and genotype of Gaucher disease in 14 children. Zhonghua Er Ke Za Zhi. (2022) 6:527–32. doi: 10.3760/cma.j.cn112140-20220228-00159
13. Hughes D, Mikosch P, Belmatoug N, Carubbi F, Cox T, Goker-Alpan O, et al. Gaucher disease in bone: from pathophysiology to practice. J Bone Miner Res. (2019) 34:996–1013. doi: 10.1002/jbmr.3734
14. Kaluzna M, Trzeciak I, Ziemnicka K, Machaczka M, Ruchała M. Endocrine and metabolic disorders in patients with gaucher disease type 1: a review. Oiphanet J Rare Dis. (2019) 14:275. doi: 10.1186/s13023-019-1211-5
15. Weinreb NJ, Goker-Alpan O, Kishnani PS, Longo N, Burrow TA, Bernat JA, et al. The diagnosis and management of gaucher disease in pediatric patients: where do we go from here? Mol Genet Metab. (2022) 136:4–21. doi: 10.1016/j.ymgme.2022.03.001
16. Schiffmann R, Sevigny J, Rolfs A, Davies EH, Goker-Alpan O, Abdelwahab M, et al. The definition of neuronopathic gaucher disease. J Inherit Metab Dis. (2020) 43:1056–9. doi: 10.1002/jimd.12235
17. Poffenberger CN, Inati S, Tayebi N, Stubblefield BK, Ryan E, Schiffmann R, et al. EEG Abnormalities in patients with chronic neuronopathic Gaucher disease: a retrospective review. Mol Genet Metab. (2020) 3:358–63. doi: 10.1016/j.ymgme.2020.10.010
18. McNeill A, Roberti G, Lascaratos G, Hughes D, Mehta A, Garway-Heath DF, et al. Retinal thinning in Gaucher disease patients and carriers: results of a pilot study. Mol Genet Metab. (2013) 109:221–3. doi: 10.1016/j.ymgme.2013.04.001
19. Kaplan P, Baris H, De Meirleir L, Di Rocco M, El-Beshlawy A, Huemer M, et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. (2013) 4:447–58. doi: 10.1007/s00431-012-1771-z
20. Kurolap A, Del Toro M, Spiegel R, Gutstein A, Shafir G, Cohen IJ, et al. Gaucher disease type 3c: new patients with unique presentations and review of the literature. Mol Genet Metab. (2019) 127:138–46. doi: 10.1016/j.ymgme.2019.05.011
21. Kor Y, Keskin M, Ba§pinar O. Severe cardiac involvement in Gaucher type IIIC: a case report and review of the literature. Cardiol Young. (2017) 27:1426–9. doi: 10.1017/S1047951117000579
22. Gawad Tantawy AA, Moneam Adly AA, Madkour SS, Salah El-Din NY. Pulmonary manifestations in young Gaucher disease patients: phenotype-genotype correlation and radiological findings. Pediatr Pulmonol. (2020) 2:441–8. doi: 10.1002/ppul.24544
23. Li D, Tao XJ, Zhang NN, Zhou ZF, Shen HW, Zhang YH, et al. Analysis of CT features of chest in Gaucher disease. Chin J Radiol. (2020) 1:23–7. doi: 10.3760/cma.j.issn.1005_1201.2020.01.005
24. Lo SM, Liu J, Chen F, Pastores GM, Knowles J, Boxer M, et al. Pulmonary vascular disease in Gaucher disease: clinical spectrum, determinants of phenotype and long-term outcomes of therapy. J Inherit Metab Dis. (2011) 34:643–50. doi: 10.1007/s10545-011-9313-9
25. Pastores GM, Hughes DA. Gaucher disease. GeneReviews®. Seattle (WA): University of Washington. 1993–2023.
26. Revel-Vilk S, Szer J, Mehta A, Zimran A. How we manage Gaucher disease in the era of choices. Br J Haematol. (2018) 182:467–80. doi: 10.1111/bjh.15402
27. Gupta P, Pastores G. Pharmacological treatment of pediatric Gaucher disease. Expert Rev Clin Pharmacol. (2018) 11:1183–94. doi: 10.1080/17512433.2018.1549486
28. Zimran A, Wajnrajch M, Hernandez B, Pastores GM. Taliglucerase alfa: safety and efficacy across 6 clinical studies in adults and children with Gaucher disease. Orphanet J Rare Dis. (2018) 13:36. doi: 10.1186/s13023-018-0776-8
29. Hughes DA, Deegan P, Giraldo P, Göker-Alpan Ö, Lau H, Lukina E, et al. Switching between enzyme replacement therapies and substrate reduction therapies in patients with Gaucher disease: data from the gaucher outcome survey (GOS). J Clin Med. (2022) 17:5158. doi: 10.3390/jcm11175158
30. Andersson HC, Charrow J, Kaplan P, Mistry P, Pastores GM, Prakash-Cheng A, et al. Individualization of long-term enzyme replacement therapy for Gaucher disease. Genet Med. (2005) 2:105–10. doi: 10.1097/01.GIM.0000153660.88672.3C
Keywords: gaucher disease, glucocerebrosidase, multidisciplinary collaboration, splenomegaly, case report
Citation: Zhu J, Sun Y, Zheng W and Wang C (2023) Case report: Multidisciplinary collaboration in diagnosis and treatment of child gaucher disease. Front. Pediatr. 11:1057574. doi: 10.3389/fped.2023.1057574
Received: 29 September 2022; Accepted: 16 March 2023;
Published: 30 March 2023.
Edited by:
Mo Wang, Children's Hospital of Chongqing Medical University, ChinaReviewed by:
Nicolina Cristina Sorrentino, Telethon Institute of Genetics and Medicine (TIGEM), Italy© 2023 Zhu, Sun, Zheng and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunlin Wang aHp3YW5nY2xAemp1LmVkdS5jbg==
Specialty Section: This article was submitted to Children and Health, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.