
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 06 February 2023
Sec. Genetics of Common and Rare Diseases
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1046731
 Jun Chen1,2,3
Jun Chen1,2,3 Jianjun Wang1,2,3Jing Gan1,2,3
Jianjun Wang1,2,3Jing Gan1,2,3 Rong Luo1,2,3
Rong Luo1,2,3 Zuozhen Yang4
Zuozhen Yang4 Mengmeng Liang4
Mengmeng Liang4 Xiaolu Chen1,2,3*
Xiaolu Chen1,2,3*
Background: Leigh syndrome (LS; OMIM: 256000) is a progressive neurodegenerative disease caused by genetic mutations resulting in mitochondrial oxidative phosphorylation defects. The prognosis is poor, with most children dying before the age of 2 years. MT-ATP6 variants are the most common mitochondrial DNA mutations in LS. MT-ATP6 variant-induced LS may trigger autoimmunity, and immunotherapy might be effective. Here, we present the first pediatric case of anti-aquaporin 4 (AQP4)–IgG-positive LS caused by an MT-ATP6 variant.
Case: A 1-year-old boy was hospitalized due to recurrent fever, cough, and developmental regression. Two months previously, he had developed reduced responses to stimulation and psychomotor retardation. After admission, his condition deteriorated and respiratory failure ensued. Magnetic resonance imaging of the brain showed symmetrical small patchy abnormal signals around the third ventricle, pons, and dorsal periaqueductal gray matter in the dorsal medulla. Laboratory tests revealed anti-AQP4–IgG antibodies. Anti-infection, immunoglobulin, and glucocorticoid therapy were administered for symptomatic treatment. Genetic testing revealed a de novo homogeneous pathogenic variant of MT-ATP6 (m.9176T > C, mutation ratio: 99.97%). The patient was diagnosed with anti-AQP4–IgG-positive LS, treated with “cocktail therapy” (vitamins B1, B2, C, and E, l-carnitine, and coenzyme Q10), and discharged after his condition improved. A literature review revealed that LS-induced mitochondrial defects can impact the immune system; hence, immunotherapy and early mitochondrial cocktail therapy may improve outcomes.
Conclusion: Anti-AQP4–IgG-positive LS is very rare. Patients with LS with the m.9176T > C variant of MT-ATP6 may be susceptible to autoimmune damage of the central nervous system. Early cocktail therapy combined with immunotherapy may improve their prognosis.
Leigh syndrome (LS), a common mitochondrial disease in infants and children, was first reported by the British neuropathologist Denis A. Leigh in 1951. The incidence of LS is 1 in 40,000 births (1, 2), and the condition invariably carries a poor prognosis. LS typically presents as an early-onset progressive disorder and is caused by defects in mitochondrial oxidative phosphorylation. Its most common clinical characteristics include feeding difficulties, growth retardation, ataxia, psychomotor retardation, dystonia, hypotonia, seizures, and respiratory disorders. The underlying pathology is a subacute necrotizing encephalomyelopathy characterized by bilateral symmetrical necrotic lesions of gray matter nuclei in the basal ganglia, diencephalon, cerebellum, or brainstem. Bilateral lesions in the basal ganglia, thalamus, brainstem (substantia nigra, oculomotor nuclei, periaqueductal gray matter), pontine tegmentum, and inferior olivary nuclei are the most common imaging findings (3). In the majority of cases, the etiology is dysfunction of the mitochondrial respiratory chain caused by either mitochondrial or nuclear gene mutations, which show considerable genetic heterogeneity. The treatment of LS essentially consists of “cocktail therapy” to modulate cell metabolism and correct mitochondrial dysfunction.
Aquaporin-4 (AQP4) is a water-transporting protein expressed in the plasma membrane of astrocytes throughout the central nervous system (CNS). AQP4 is widely involved in diverse functions, such as the regulation of cerebrospinal fluid (CSF) circulation, interstitial fluid resorption, Ca(2+) signaling, potassium buffering, neuroexcitation, and neuroinflammation (4). Therefore, AQP4 is thought to be closely associated with epilepsy, cerebral edema, stroke, multiple sclerosis, neuromyelitis optica spectrum disorder (NMOSD), post-traumatic brain injury, Parkinson disease, and other CNS disorders. AQP4–IgG is generally considered to be the specific autoantibody of NMOSD, which is a rare inflammatory CNS disease that primarily manifests as relapsing episodes of severe optic neuritis and myelitis (5). In this study, we present a very rare case of anti-AQP4–IgG-positive LS with the m.9176T > C variant of the MT-ATP6 gene. The patient exhibited an autoimmune reaction against the CNS, and immunotherapy improved the clinical symptoms.
A 15-month-old boy was admitted to our hospital due to apathetic facial expressions since 2 months, recurrent cough since 1 month, and developmental regression since half a month. Two months previously (at the age of 13 months), the patient appeared to show a lack of proper responses to stimulations, including dull eyes, decreased crying and laughing, and decreased verbal communication with his parents. Then, he developed a monophonic cough accompanied by a slight fever and runny nose. Half a month ago (at the age of 14 months), he exhibited a gradual regression in motor function, and was unable to walk or sit steadily. He was also unable to express himself and could only speak in one- or two-word responses compared with two- to three-word phrases before. The patient's milk intake had decreased from a feed of 150 ml to a feed of 30–40 ml. The patient had no history of other illnesses and no family history of genetic diseases, CNS diseases, autoimmune diseases, developmental delay, or other chronic diseases. A physical examination showed that the patient was awake with apathetic facial expressions. A chest examination revealed rough breath sounds without rales or wheezing. The patient could raise his hands and legs but could not stand or walk steadily due to hypotonia. The Babinski sign was positive on both sides, and the meningeal irritation sign was negative. Laboratory tests performed on admission showed the following: white blood cell count, 19.21 × 109/L (normal range, 4–9 × 109/L); procalcitonin, 0.74 ng/ml (normal range, <0.15 ng/ml); and blood lactate, 4.2 mmol/L (normal range, 0.5–1.7 mmol/L) (Figure 1). Blood cultures were positive for Staphylococcus hominis subsp., which was susceptible to vancomycin and levofloxacin. A CSF examination revealed a lactate level of 3.5 mmol/L (normal range, 0.999–2.775 mmol/L); the results of CSF cytology, biochemistry, and culture examinations were all normal. Cell-based assays were performed to assess the serum levels of anti-AQP4–IgG, anti-myelin oligodendrocyte glycoprotein, anti-glial fibrillary acidic protein, and anti-myelin basic protein antibodies: only the serum anti-AQP4–IgG test was positive (Figure 2), with a titer of 1:10 (initial titer, 1:1). Identical oligoclonal bands in the serum and CSF were observed (“mirror pattern,” type 4). Antibodies associated with autoimmune encephalitis were not detected. The auditory brainstem response test showed bilateral prolongation of wave V, and III–V and I–V interwave latencies. The wave amplitudes were decreased. Visual evoked potential testing showed that the P100 wave had a lower amplitude and a latency delay in both eyes. Cranial magnetic resonance imaging (MRI) revealed bilateral symmetrical lesions of abnormal signal intensity around the third ventricle, pons, dorsal medulla oblongata, and in the gray matter around the dorsal aqueduct on T2-weighted images and fluid attenuated inversion recovery (FLAIR) images (Figure 3). Spinal MRI showed no abnormalities in the cervical, thoracic, and lumbar spine. A chest x-ray revealed bilateral pneumonia. An antibiotic treatment regimen of vancomycin was initiated according to the identified bacteria (Staphylococcus hominis subsp.) and drug-sensitivity results.
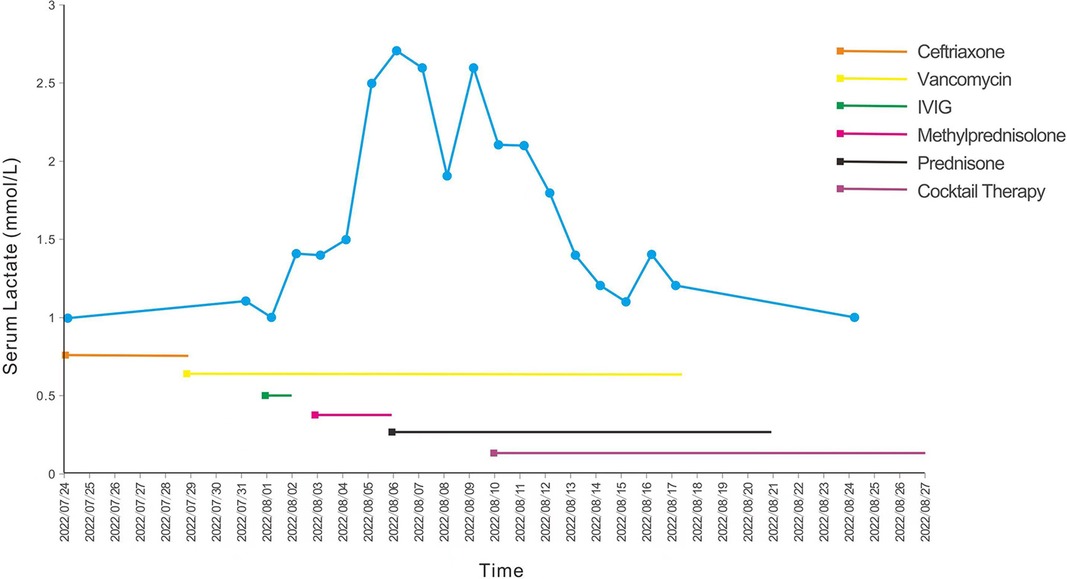
Figure 1. Blood lactate curve and treatment of the patient. The blue line represents the blood lactate levels of the patient after admission, and the straight lines with different colors represent different therapeutic drugs. IVIG, intravenous immunoglobulin.
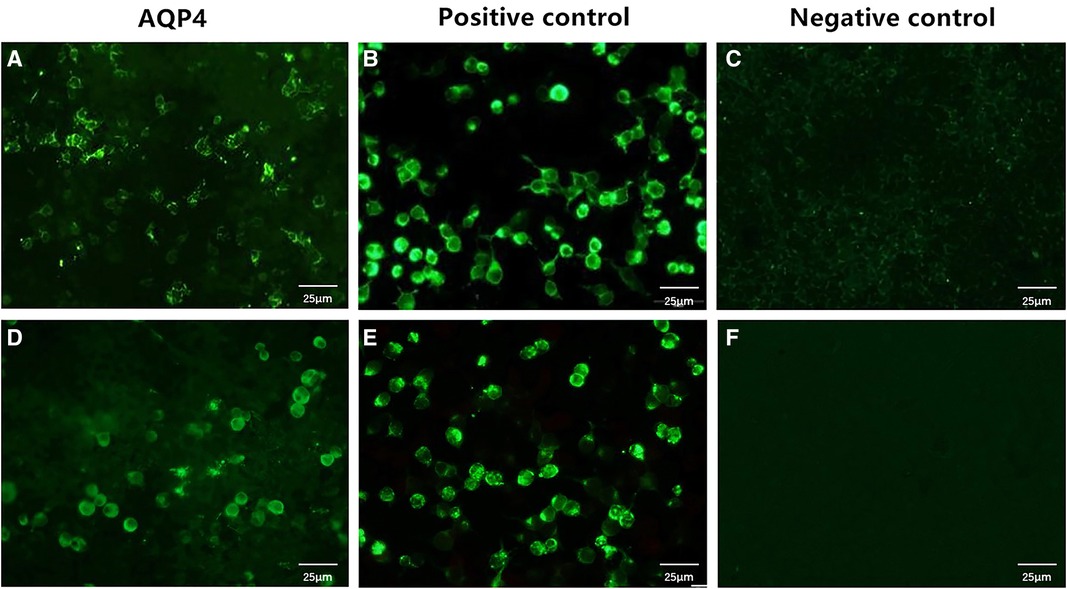
Figure 2. Serum anti-AQP4–IgG levels in the patient. The serum samples of the patient were examined using cell-based assays, which used HEK293 cells transfected with human AQP4. The anti-AQP4 antibody test is positive (A), and the antibody titer is 1:10 (initial titer, 1:1). After 3 days, the anti-AQP4 antibody test is still positive, with an antibody titer of 1:10 (D). (B,E) are positive controls, while (C,F) are negative controls. Cells stained green are positive for the anti-AQP4 antibody; unstained cells are negative for this antibody. Scale bar: 25 μm. AQP4, aquaporin-4.
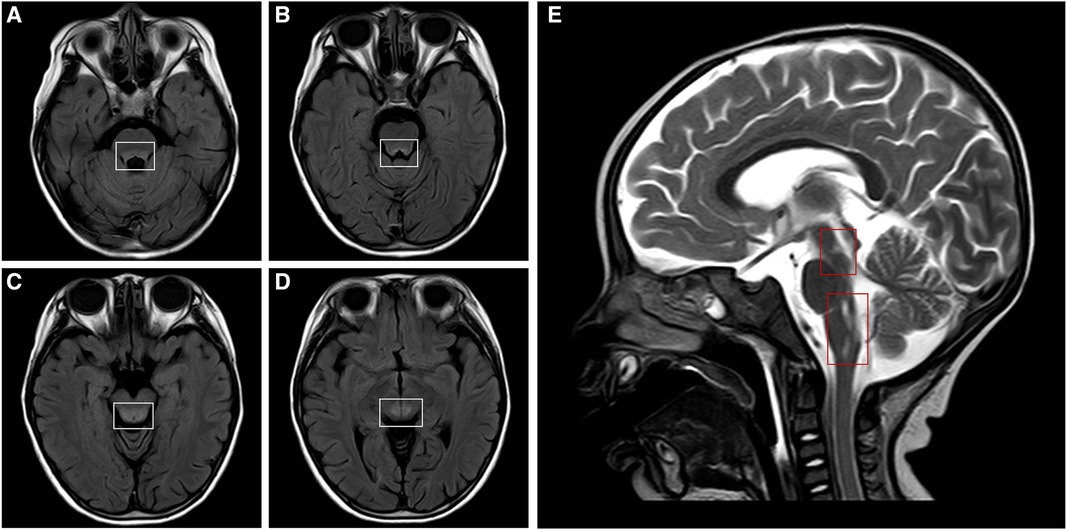
Figure 3. Cranial MRI of the patient. Cranial MRI shows bilateral symmetrical lesions of abnormal signal intensity around the third ventricle, pons, dorsal medulla oblongata, and in the gray matter around the dorsal aqueduct. Axial T2-FLAIR images (A–D, white frames) and a sagittal T2-weighted image (E, red frames) show the lesions. MRI, magnetic resonance imaging; FLAIR, fluid attenuated inversion recovery.
On day 7 of admission, the condition of the patient worsened; he developed drowsiness, shallow irregular breathing, hypotonia, and fever, which necessitated mechanical ventilation. The patient was transferred to the pediatric intensive care unit due to respiratory failure and unconsciousness (Glasgow coma scale, 10), and was administered invasive mechanical ventilation. Due to the positive anti-AQP4–IgG antibody test, abnormal visual evoked potentials, and abnormal signals on brain MRI, the possibility of NMOSD could not be excluded.
The NMOSD diagnostic criteria issued by the International Panel for Neuromyelitis Optica Diagnosis in 2015 (6) are as follows: (1) patients must have experienced at least one of the six core clinical characteristics of NMOSD, namely, optic neuritis, acute myelitis, area postrema syndrome, acute brainstem syndrome, symptomatic narcolepsy or acute diencephalic clinical syndrome along with NMOSD-typical diencephalic MRI lesions, or symptomatic cerebral syndrome along with NMOSD-typical brain lesions; (2) a positive test for AQP4–IgG (using the best available detection method, such as a cell-based assay); and (3) the exclusion of alternative diagnoses. Our patient had no core clinical characteristics of NMOSD but tested positive for anti-AQP4–IgG. To rule out false-positive results, we reexamined the anti-AQP4–IgG titer 3 days later, and the result was still positive (titer, 1 : 10) (Figure 2).
Although the patient did not meet all the diagnostic criteria for NMOSD, considering that AQP4–IgG is a pathogenic antibody and that autoimmune brain injury could not be completely ruled out, we treated the patient with intravenous immunoglobulin (1 g/kg/day, daily) for 2 days (on days 8 and 9 after admission) and a 3-day course (on days 10–13 after admission) of intravenous methylprednisolone (20 mg/kg/day), followed by oral prednisone (2 mg/kg/day) for 2 weeks (on days 14–28 after admission). After the above treatment, the clinical signs and symptoms of the patient improved, and on day 17 after admission, he was transferred to the pediatric neurology ward for further treatment. Owing to the patient's medical history, clinical manifestations, hyperlactatemia (in serum and CSF), and cranial MRI results, we also considered a diagnosis of the mitochondrial disease LS. Bilateral pneumonia may lead to the deterioration of the primary disease. Therefore, on day 17 after admission, we initiated persistent “cocktail therapy” that included coenzyme Q10 (10 mg/kg/day), l-carnitine (50 mg/kg/day), and multiple vitamins (vitamin B1, vitamin B2, vitamin E, and vitamin C). We also collected peripheral blood samples from the patient and his parents for genetic testing. We detected a de novo homogeneous variant in the MT-ATP6 gene: m.9176T > C (mutation ratio, 99.97%) (Figure 4). This variant is one of the most common variants in this mitochondrial disorder and has been reported as a pathogenic variant in the ClinVar database.
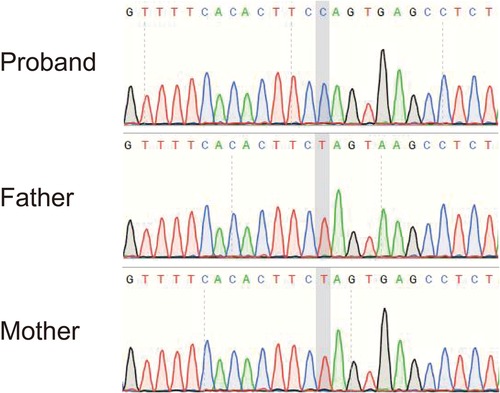
Figure 4. Sequencing result of the MT-ATP6 gene in the patient. The sequencing analysis revealed a de novo homogeneous variant (m.9176T > C) in the MT-ATP6 gene, which is one of the most common variants in Leigh syndrome.
After the diagnosis of LS, we continued to administer the cocktail therapy to the patient. His condition improved, and his serum lactate level decreased (Figure 1); however, he had significant retardation with delayed motor and language development. He also needed to be fed through a gastric tube due to difficulty in swallowing. After a total of 5 weeks in hospital, the patient was finally discharged back home. After being discharged, the patient has continued rehabilitative therapy, and we have suggested a follow-up examination in the outpatient clinic after 3 months (which is yet to occur at the time of writing).
The onset of LS usually occurs in early infancy or early childhood, and the psychomotor development is usually normal before onset. The main manifestations of LS include psychomotor regression, dystonia, feeding difficulties, ataxia, and brainstem dysfunction (abnormal breathing, ophthalmoplegia, nystagmus, and recurrent vomiting), which are accompanied by increased lactic acid levels in the blood, CSF, and urine (7). The onset of LS is typically induced by a trigger, such as an acute infection or surgery. After the onset of symptoms, the disease advances rapidly, with most patients succumbing to the disease due to respiratory failure within 2 years. The clinical diagnosis of LS requires the following three characteristics (8): (1) neurodegenerative diseases with various clinical manifestations; (2) symmetrical hyperintensities in the basal ganglia and/or brainstem on T2-weighted imaging, and a lactate peak in the affected areas on spectroscopy; and (3) mitochondrial dysfunction caused by a gene mutation.
In this case, the patient developed symptoms at the age of 1 year, and his development was normal before the onset of symptoms. His initial symptoms involved mental status and behavioral changes (such as apathy and a decline in conversation), poor language production, and retardation of motor functions and dystonia, accompanied by elevated lactate levels in the blood and CSF. The imaging findings included bilateral symmetrical hyperintense signal abnormalities in the brainstem and basal ganglia on T2-weighted MRI, which are typical for LS. Genetic tests detected a de novo homogeneous pathogenic variant in the MT-ATP6 gene, namely, m.9176T > C. Thus, the diagnosis of LS was clear in this patient. After the onset of symptoms, the patient's condition rapidly progressed to disorders of consciousness and respiratory failure, necessitating mechanical ventilation.
To date, more than 60 genetic mutations have been reported as causative mutations of LS, of which 70%–80% are nuclear DNA mutations and 10%–20% are mitochondrial DNA (mtDNA) mutations. The mtDNA gene MT-ATP6 (OMIM: 516,060), which encodes the ATPase6 subunit of the mitochondrial ATP synthase, is located at mtDNA 8527–9207. In this case of LS, the m.9176T > C variant was identified in the MT-ATP6 gene. To date, 16 cases of LS with the m.9176T > C variant have been reported (Table 1) (9–18). Among these 16 patients, 6 patients were treated with cocktail therapy, of whom 5 patients have survived to date and 1 patient has died (18). In the patient who died, cocktail therapy was begun 2 months after the onset of symptoms, which suggests that early mitochondrial cocktail therapy may improve patients’ outcomes.
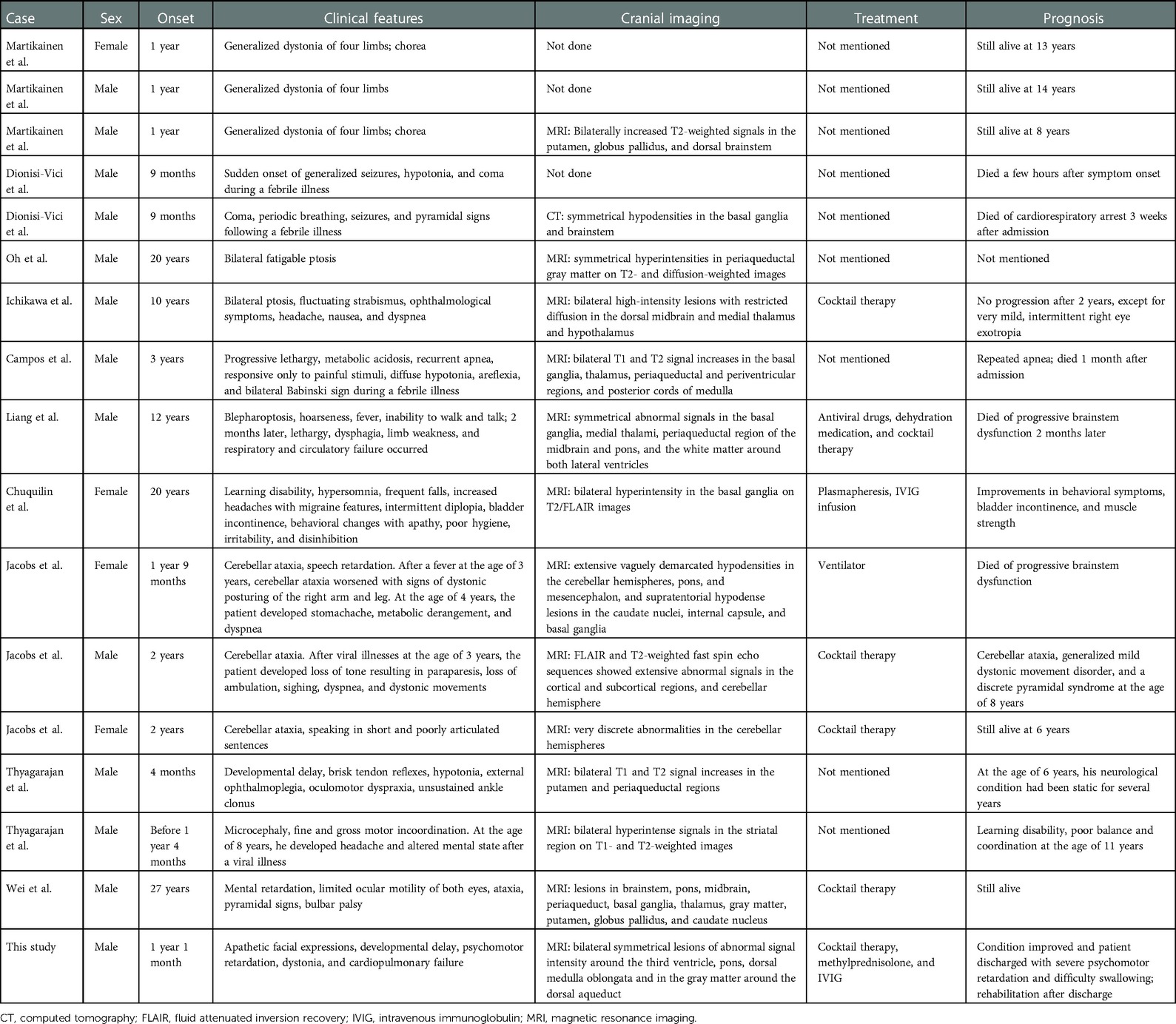
Table 1. Cases of Leigh syndrome with the T9176C variant of the MT-ATP6 gene reported in the literature.
The m.9176T > C variant of the MT-ATP6 gene results in the replacement of a highly conserved leucine residue (amino acid 270) with a proline residue, which decreases the synthesis of mitochondrial proteins and reduces the activity of oxidative enzymes, causing a decrease in mitochondrial ATP synthesis (19). The reduced ATP production increases the mitochondrial transmembrane potential (with resultant hyperpolarization) and reactive oxygen species production, which in turn activates the necroptosis pathway, resulting in the release of sequestered autoantigens and enhancing the immunogenic properties of neural cells (20). The above findings suggest that secondary cellular necrosis may result in a potential immunoinflammatory reaction in patients with LS with the m.9176T > C variant of the MT-ATP6 gene.
Our patient also tested positive for serum anti-AQP4–IgG antibodies. In addition, oligoclonal bands were detected in both the serum and CSF of the patient, and his condition improved after intravenous immunoglobulin and glucocorticoid treatment. AQP4–IgG is associated with NMOSD in the majority of cases and has been demonstrated to be directly pathogenic and of high differential diagnostic and prognostic impact (21, 22). The reason for the co-occurrence of LS and anti-AQP4–IgG antibodies in this patient is currently unclear. The AQP4 protein is abundantly expressed in the foot processes of astrocytes that are adjacent to the blood vessels and piamater (around the blood–brain barrier) (23), which is a common site of involvement in LS. A recent study found that mitochondrial dysfunction in the CNS and the peripheral blood may contribute to disease progression and the disability level in patients with NMOSD (24). In patients with LS, mitochondrial dysfunction leads to cell edema and hypoxia, which results in the loss of energy-dependent solute homeostasis and drives water influx down an osmotic gradient into perivascular astrocytes, which ultimately causes swelling (25). Therefore, we speculated that the presence of AQP4 antibodies may be related to the release of autoimmune substances or the exposure of the AQP4 surface antigen secondary to astrocyte edema and necrosis caused by mitochondrial dysfunction. The binding of IgG to AQP4 can lead to the generation of reactive astrocytes and the early recruitment of granulocytes, leading to the amplification of the inflammatory response, reversible internalization of AQP4, direct impairment of water flux independent of antigen downregulation, and downregulation of excitatory amino acid transporter 2, with consequent glutamate excitotoxicity, and complement-dependent and antibody-dependent cellular cytotoxicity that can also damage surrounding cells (5, 26). Increased AQP4–IgG may also worsen mitochondrial dysfunction associated with Na+-K+-ATPase, which could result in increased intracellular crystalloid osmotic pressure and cytotoxic edema.
There is growing evidence that mitochondrial dysfunction can impact every organ system, including the immune system. A recently published study demonstrated that targeting immune cells through high-dose leukocyte-specific inhibitors rescued CNS degeneration in diseases such as LS (27). In addition, plasma exchange synchronized with intravenous gamma globulin therapy was reported to be effective in one patient with LS who also had the m.9176T > C variant of the MT-ATP6 gene (15). The authors of that case report considered that metabolic damage to neuronal cells may trigger an autoimmune response in mitochondrial diseases. The authors also pointed out that oligoclonal bands have been detected in mitochondrial diseases such as Leber hereditary optic neuropathy, progressive external ophthalmoplegia associated with POLG gene mutation mimicking multiple sclerosis, and acute demyelinating encephalomyelitis (28, 29). In one case report, a patient with a confirmed mitochondrial myopathy was found to have muscle T-cell infiltration upon biopsy and showed significant clinical improvement after intravenous immunoglobulin therapy; this case supports the idea of a pathological inflammatory response induced by mitochondrial diseases (30). Mancuso et al. (30) also reported that excellent results were achieved in a patient with mitochondrial myopathy after intravenous immunoglobulin and carnitine treatment, suggesting that mitochondrial diseases might cause a pathological autoimmune inflammatory reaction in the CNS.
Although none of the discussed preclinical therapeutics or clinical immune-targeting therapeutics are approved to treat genetic mitochondrial diseases, corticosteroids can provide significant and at times persistent benefits in genetically and clinically distinct forms of mitochondrial diseases, including MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes), mitochondrial myopathy, mitochondrial encephalomyopathy, mitochondrial neurogastrointestinal encephalopathy, and mitochondrial leukoencephalopathy (31). Mechanistic target of rapamycin (mTOR) inhibitors, rapamycins, and analogs, such as sirolimus and everolimus, have immunosuppressive actions that are mechanistically distinct from corticosteroids, and these have also been reported to have therapeutic effects in mitochondrial diseases. A 2-year-old patient with LS responded well to mTOR inhibition therapy, with a striking reversal of gross motor function loss and brain lesions, which persisted through 20 months of therapy (32). These findings have dramatically changed our understanding of the pathogenesis of LS, demonstrating that immune involvement is causal in mitochondrial diseases. Further work is needed to elucidate the precise mechanisms between mitochondrial dysfunction, immune dysregulation, and pathology.
In this study, we have reported a very rare case of anti-AQP4–IgG-positive LS caused by a variant of MT-ATP6. Mitochondrial diseases can trigger an autoimmune inflammatory reaction, which influences many organs, especially the CNS. Although previous studies and the present case show that immunotherapy may be effective for the treatment of LS with the m.9176T > C variant, further studies are needed to reveal the intrinsic mechanisms underlying LS with the m.9176T > C variant, which is prone to cause autoimmune injury in the CNS. More cases are needed to verify the efficacy of immunotherapy in this LS subtype.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by West China Second University Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
JC: conceptualization, methodology, and writing original draft. JW, JG, and RL: methodology and review. ZY and ML: software, data mining, and investigation. XC: supervision, writing, reviewing, and editing. All authors contributed to the article and approved the submitted version.
This work was supported by the Popularization and Application Project of Sichuan Provincial Health Commission (21PJ049) and grants from the research funds of West China Second University Hospital (No. KL115, KL072).
We would like to thank the patient and his family for their cooperation.
ZY and ML were employed by Cipher Gene LLC.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AQP4, aquaporin-4; CNS, central nervous system; CSF, cerebrospinal fluid; CT, computed tomography; FLAIR, fluid attenuated inversion recovery; IVIG, intravenous immunoglobulin; LS, Leigh syndrome; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; MRI, magnetic resonance imaging; mTOR, mechanistic target of rapamycin; mtDNA, mitochondrial DNA; NMOSD, neuromyelitis optica spectrum disorder.
1. Leigh D. Subacute necrotizing encephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry. (1951) 14(3):216–21. doi: 10.1136/jnnp.14.3.216
2. Darin N, Oldfors A, Moslemi AR, Holme E, Tulinius M. The incidence of mitochondrial encephalomyopathies in childhood: clinical features and morphological, biochemical, and DNA abnormalities. Ann Neurol. (2001) 49(3):377–83. doi: 10.1002/ana.75
3. Lake NJ, Bird MJ, Isohanni P, Paetau A. Leigh syndrome: neuropathology and pathogenesis. J Neuropathol Exp Neurol. (2015) 74(6):482–92. doi: 10.1097/NEN.0000000000000195
4. Verkman AS, Smith AJ, Phuan PW, Tradtrantip L, Anderson MO. The aquaporin-4 water channel as a potential drug target in neurological disorders. Expert Opin Ther Targets. (2017) 21(12):1161–70. doi: 10.1080/14728222.2017.1398236
5. Pittock SJ, Zekeridou A, Weinshenker BG. Hope for patients with neuromyelitis optica spectrum disorders—from mechanisms to trials. Nat Rev Neurol. (2021) 17(12):759–73. doi: 10.1038/s41582-021-00568-8
6. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. (2015) 85(2):177–89. doi: 10.1212/WNL.0000000000001729
7. Gerards M, Sallevelt SC, Smeets HJ. Leigh syndrome: resolving the clinical and genetic heterogeneity paves the way for treatment options. Mol Genet Metab. (2016) 117(3):300–12. doi: 10.1016/j.ymgme.2015.12.004
8. Baertling F, Rodenburg RJ, Schaper J, Smeitink JA, Koopman WJ, Mayatepek E, et al. A guide to diagnosis and treatment of Leigh syndrome. J Neurol Neurosurg Psychiatry. (2014) 85(3):257–65. doi: 10.1136/jnnp-2012-304426
9. Wei Y, Cui L, Peng B. Mitochondrial DNA mutations in late-onset Leigh syndrome. J Neurol. (2018) 265(10):2388–95. doi: 10.1007/s00415-018-9014-5
10. Martikainen MH, Ng YS, Gorman GS, Alston CL, Blakely EL, Schaefer AM, et al. Clinical, genetic, and radiological features of extrapyramidal movement disorders in mitochondrial disease. JAMA Neurol. (2016) 73(6):668–74. doi: 10.1001/jamaneurol.2016.0355
11. Dionisi-Vici C, Seneca S, Zeviani M, Fariello G, Rimoldi M, Bertini E, et al. Fulminant Leigh syndrome and sudden unexpected death in a family with the T9176c mutation of the mitochondrial atpase 6 gene. J Inherit Metab Dis. (1998) 21(1):2–8. doi: 10.1023/a:1005397227996
12. Oh EH, Chae SH, Cho JW, Baik SK, Choi SY, Choi KD, et al. Fatigable ptosis as an initial presentation of adult-onset Leigh syndrome. Neurology. (2017) 89(16):1754. doi: 10.1212/WNL.0000000000004528
13. Ichikawa K, Tsuyusaki Y, Shimbo H, Goto T. Late-onset Leigh syndrome with M.9176t > C mutation in the mitochondrial atpase 6 gene. Pediatr Int. (2019) 61(10):1055–6. doi: 10.1111/ped.13991
14. Campos Y, Martin MA, Rubio JC, Solana LG, Garcia-Benayas C, Terradas JL, et al. Leigh syndrome associated with the T9176c mutation in the atpase 6 gene of mitochondrial DNA. Neurology. (1997) 49(2):595–7. doi: 10.1212/wnl.49.2.595
15. Chuquilin M, Govindarajan R, Peck D, Font-Montgomery E. Response to immunotherapy in a patient with adult onset Leigh syndrome and T9176c Mtdna mutation. Mol Genet Metab Rep. (2016) 8:28–32. doi: 10.1016/j.ymgmr.2016.06.004
16. Jacobs LJ, de Coo IF, Nijland JG, Galjaard RJ, Los FJ, Schoonderwoerd K, et al. Transmission and prenatal diagnosis of the T9176c mitochondrial DNA mutation. Mol Hum Reprod. (2005) 11(3):223–8. doi: 10.1093/molehr/gah152
17. Thyagarajan D, Shanske S, Vazquez-Memije M, De Vivo D, DiMauro S. A novel mitochondrial atpase 6 point mutation in familial bilateral striatal necrosis. Ann Neurol. (1995) 38(3):468–72. doi: 10.1002/ana.410380321
18. Liang JM, Xin CJ, Wang GL, Wu XM. Late-onset Leigh syndrome without delayed development in China: a case report. World J Clin Cases. (2021) 9(24):7133–8. doi: 10.12998/wjcc.v9.i24.7133
19. Kucharczyk R, Ezkurdia N, Couplan E, Procaccio V, Ackerman SH, Blondel M, et al. Consequences of the pathogenic T9176c mutation of human mitochondrial DNA on yeast mitochondrial Atp synthase. Biochim Biophys Acta. (2010) 1797(6–7):1105–12. doi: 10.1016/j.bbabio.2009.12.022
20. Chuquilin M, Govindarajan R, Peck D, Font Montgomery E. Response to letter to the editor: why does Leigh syndrome responds to immunotherapy? Mol Genet Metab Rep. (2016) 8:85–6. doi: 10.1016/j.ymgmr.2016.08.003
21. Jarius S, Aboul-Enein F, Waters P, Kuenz B, Hauser A, Berger T, et al. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain. (2008) 131(Pt 11):3072–80. doi: 10.1093/brain/awn240
22. Jarius S, Wildemann B, Paul F. Neuromyelitis optica: clinical features, immunopathogenesis and treatment. Clin Exp Immunol. (2014) 176(2):149–64. doi: 10.1111/cei.12271
23. Saini H, Fernandez G, Kerr D, Levy M. Differential expression of aquaporin-4 isoforms localizes with neuromyelitis optica disease activity. J Neuroimmunol. (2010) 221(1–2):68–72. doi: 10.1016/j.jneuroim.2010.02.007
24. Foolad F, Khodagholi F, Nabavi SM, Javan M. Changes in mitochondrial function in patients with neuromyelitis optica; correlations with motor and cognitive disabilities. PLoS One. (2020) 15(3):e0230691. doi: 10.1371/journal.pone.0230691
25. Bordone MP, Salman MM, Titus HE, Amini E, Andersen JV, Chakraborti B, et al. The energetic brain—a review from students to students. J Neurochem. (2019) 151(2):139–65. doi: 10.1111/jnc.14829
26. Lerch M, Schanda K, Lafon E, Wurzner R, Mariotto S, Dinoto A, et al. More efficient complement activation by anti-aquaporin-4 compared with anti-myelin oligodendrocyte glycoprotein antibodies. Neurol Neuroimmunol Neuroinflamm. (2023) 10(1):e200059. doi: 10.1212/NXI.0000000000200059
27. Stokes JC, Bornstein RL, James K, Park KY, Spencer KA, Vo K, et al. Leukocytes mediate disease pathogenesis in the Ndufs4(Ko) mouse model of Leigh syndrome. JCI Insight. (2022) 7(5):e156522. doi: 10.1172/jci.insight.156522
28. Echaniz-Laguna A, Chassagne M, de Seze J, Mohr M, Clerc-Renaud P, Tranchant C, et al. Polg1 variations presenting as multiple sclerosis. Arch Neurol. (2010) 67(9):1140–3. doi: 10.1001/archneurol.2010.219
29. Slee M, Finkemeyer J, Krupa M, Raghupathi R, Gardner J, Blumbergs P, et al. A novel mitochondrial DNA deletion producing progressive external ophthalmoplegia associated with multiple sclerosis. J Clin Neurosci. (2011) 18(10):1318–24. doi: 10.1016/j.jocn.2011.02.019
30. Mancuso M, Orsucci D, Ienco EC, Ricci G, Ali G, Servadio A, et al. An “inflammatory” mitochondrial myopathy. A case report. Neuromuscul Disord. (2013) 23(11):907–10. doi: 10.1016/j.nmd.2013.07.011
31. Hanaford A, Johnson SC. The immune system as a driver of mitochondrial disease pathogenesis: a review of evidence. Orphanet J Rare Dis. (2022) 17(1):335–47. doi: 10.1186/s13023-022-02495-3
Keywords: Leigh syndrome, anti-AQP4–IgG, MT-ATP6, m.9176T > C, case report
Citation: Chen J, Wang J, Gan J, Luo R, Yang Z, Liang M and Chen X (2023) Anti-AQP4–IgG-positive Leigh syndrome: A case report and review of the literature. Front. Pediatr. 11:1046731. doi: 10.3389/fped.2023.1046731
Received: 17 September 2022; Accepted: 9 January 2023;
Published: 6 February 2023.
Edited by:
Erika Fernandez-Vizarra, University of Padua, ItalyReviewed by:
Friedemann Paul, Charité Universitätsmedizin Berlin, Germany© 2023 Chen, Wang, Gan, Luo, Yang, Liang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaolu Chen Y3hsbmpAcXEuY29t
Specialty Section: This article was submitted to Genetics of Common and Rare Diseases, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.