 Liang Wang
Liang Wang Sen Chen2
Sen Chen2
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 17 October 2022
Sec. Pediatric Hematology and Hematological Malignancies
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.993952
Acute myeloid leukemia (AML) is a clinically, morphologically, and genetically heterogeneous group of malignancies characterized by a wide range of genomic alterations responsible for defective regulation of the differentiation and self-renewal programs of hematopoietic stem cells. Here, we report a 4-month-old boy who had acute onset with leukocytosis and abdominal mass. The morphological analysis of bone marrow (BM) smear revealed extremely marrow hyperplasia, large quantities of immature cells, and primary and immature monocytic hyperplasia accounting for 57.5% of nucleated cells. The chromosome karyotype of the case was complex, representing 48, XY, +13, +19[12]/48, idem, del (p12)[8]. After RNAs sequencing, a mutation (c.346G > A, p.G116S) of the GNB1 gene was detected and localized to the mutational hotspot in Exon 7. Meanwhile, the other three mutations were identified by next-generation sequencing (NGS) and whole-exome sequencing (WES) of DNA from the BM aspirate and oral swab, including BCORL1 mutation [c.2632A > G, p.S878G, mutation allele frequency (VAF): 99.95%], SH2B3 mutation (c.1606G > A, p.A536T, VAF: 51.17%), and KMT2D mutation (c.11124C > G, p.S3708R, VAF: 48.95%). BCORL1 mutations have been associated with the pathogenesis of AML, whereas other mutations have rarely been previously reported in pediatric AML. The patient did not undergo the combination chemotherapy and eventually died of respiratory failure. In conclusion, the concurrence of BCORL1, GNB1, SH2B3, and KMT2D mutations may be a mutationally detrimental combination and contribute to disease progression.
Acute myeloid leukemia (AML) is a complex and dynamic disease characterized by high molecular heterogeneity, representing 15%–20% of leukemias in children (1). The development of cytogenetic and molecular biological techniques in recent years has significantly contributed to a better understanding of the molecular and genomic landscapes of AML. A number of genetic mutations have been identified in pediatric AML, which are related to biological, clinical, and prognostic implications. These genes included RUNX1, NPM1, CEBPA, FLT3, MYST3, EVI1, KIT, IDH1 and IDH2, DDX41, ETV6, GATA2, TP53, and BCOR/BCORL1 (2, 3). The BCL6 corepressor (BCOR) gene, and its homolog, the BCL6 corepressor-like 1 (BCORL1) mutations, mostly represented frameshifts (deletions, insertions), nonsense and missense mutations, which led to the lack or low expression of the protein. BCORL1 mutations were detected in 3.7%–5.8% of AML adult patients and 1.2% of pediatric AML patients (4).
Mutations in guanine nucleotide-binding protein (G protein), and beta polypeptide 1 (GNB1) have been linked to neurodevelopmental syndrome (5). SH2B adaptor protein 3 (SH2B3) mutations have been identified in a range of hematological diseases, including myeloproliferative neoplasms (MPN), myelodysplastic syndromes (MDS), MDS/MPN overlap syndromes, and acute lymphoblastic leukemia (ALL) (6). Mutations in lysine-specific methyltransferase 2D (KTM2D) were the predominant cause of diffuse large B-cell lymphoma and follicular lymphoma (7). GNB1, KMT2D, and SH2B3 mutations were rarely described in pediatric AML. We reported a pediatric AML, while rare, bearing BCORL1, GNB1, KMT2D, and SH2B3 tetramutation and reviewed the relevant literature and cases of AML to improve the understanding of this rare form of pediatric AML.
A 4-month-old boy presented to the hospital in June 2021 with a high-grade fever lasting more than 6 days and diarrhea, and a CBC showing anemia (HGB 97 g/L) and leukocytosis (WBC 91.57 × 109/L). Physical examination upon admission showed an absence of spleen/liver enlargement or cutaneous lesions but the existence of abdominal mass. A chest x-Rry was performed, showing the inflammatory consolidation of the lungs. The patient was admitted to the hospital with a suspicion of leukemia. After admission, follow-up blood tests confirmed the presence of moderate anemia (HGB 83 g/L, MCV 86.7 fl, MCH 26.9 pg, MCHC 311 g/L) and leukocytosis (WBC 116.44 × 109/L) with 60% blasts on the peripheral blood smear, as well as the elevated d-dimer (1.11 mg/L), Cr (59 µmol/L), UA (487 µmol/L), GGT (788 U/L), AST (111 U/L), LDH (1,830 U/L), and CRP (19.8 mg/L). In addition, blood, urine, and stool culture were all negative. The examinations of the cerebrospinal fluid showed no significant abnormalities. An examination of the patient’s bone marrow (BM) smear revealed extremely marrow hyperplasia, large quantities of immature cells, primary and immature monocytic hyperplasia accounting for 57.5% of 200 nucleated cells, hypoplasia in the granulocyte and erythrocyte series [granulocyte series accounting for 25.5% (reference range: 40%–60%) and erythrocyte series accounting for 3% (reference range: 15%–25%)] and lymphopenia. Immunohistochemistry showed: leukemic cells HLA-DR(+), CD4(+), CD7(+), CD33(+), CD34(+), CD117(+), CD123(+). The immunophenotyping and morphological analysis on BM aspirate showed suggestive AML-M5.
Cytogenetic analysis revealed that the chromosome karyotype was complex, representing 48, XY, +13, +19[12]/48, idem, del(p12)[8] (Figure 1). FISH studies and multiplex leukemia translocation assay were negative for common fusion translocations in AML. After RNAs sequencing was performed with the Illumina HiSeq X platform, a mutation (p.G116S) of the GNB1 gene was detected, which localized to the mutational hotspot in Exon 7. The clinical and/or experimental evidence of genomic variants were assessed by the Association for Molecular Pathology (AMP), American Society of Clinical Oncology (ASCO), and College of American Pathologists (CAP) proposed Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer (8). Sequence variants were categorized into four categories based on their clinical impact: tier I, variants with strong clinical significance (level A and B evidence); tier II, variants with potential clinical significance (level C or D evidence); tier III, variants with unknown clinical significance; and tier IV, variants that are benign or likely benign. The variant of GNB1 (G116S) in this report was categorized into tier II, which may be associated with the pathogenesis of AML.
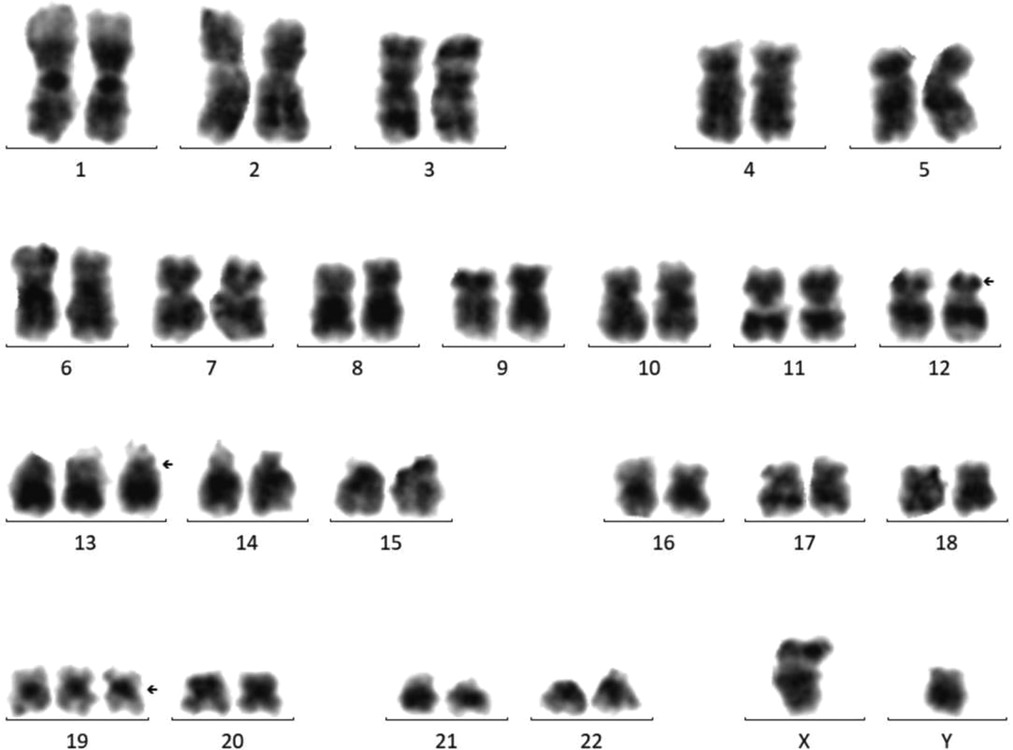
Figure 1. Bone marrow karyotype showing 48, XY, +13, +19[12]/48, idem, del(p12)[8].
Furthermore, a next-generation sequencing (NGS)-based analysis and whole-exome sequencing (WES) for the detection of mutations were performed on the DNAs extracted from BM samples and oral swabs containing exfoliated cells. The three mutations were detected, including BCORL1 mutation (p.S878G), SH2B3 mutation (p.A536T), and KMT2D mutation (p.S3708R), and it was confirmed that these mutations were germline variants (Table 1). We determined the pathogenicity of our variants via the rules for combining criteria to classify sequence variants claimed in 2015 the American College of Medical Genetics and Genomics (ACMG) and AMP proposed Standards and Guidelines for the Interpretation of Sequence Variants (2015 ACMG/AMP guidelines) (9). Variants were classified as pathogenic, likely pathogenic, uncertain significance (VUS), likely benign, or benign. As per ACMG classification, the variants of SH2B3 and KMT2D may be VUS, but the variants of BCORL1 may be likely benign. There is no sufficient literature to support pathological significance, and with further research, these variant sites may have relevant pathological significance in the future.
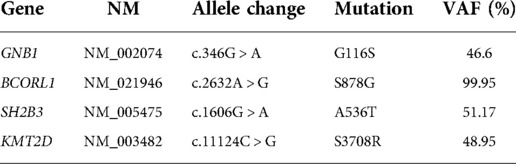
Table 1. The mutations of pediatric AML in this study.
The patient presented with pneumonia and was administered intravenous latamoxef sodium (40 mg/kg/day, every 12 h) and subsequently meropenem (20 mg/kg/day, every 8 h) for anti-infection therapies. Several other symptomatic supportive treatments were used to prevent hyperuricemia and edema. Because the relatives disagreed with the combination chemotherapy, the patient did not receive medication. After 26 days of hospitalization, the patient eventually died of respiratory failure.
PubMed database was searched with keywords including acute myeloid leukemia, BCORL1, GNB1, SH2B3, and KMT2D. It has been reported that 26 different types of BCORL1 mutations were described (2, 10–13) (Figure 2A). Pediatric AML with BCORL1 mutations is very rare. To our knowledge, only six cases have been reported in the literature (2, 13). Most of them had complex chromosomal karyotypes with diverse chromosomal abnormalities (Table 2). Only one patient with SH2B3 mutation (p.S417*) in SH2 domains was reported in a cohort of 1,540 patients with AML till now (14). GNB1 mutations were present in 3 (1.9%) of 157 cases of myelodysplastic syndrome (MDS) or secondary AML (15). However, KMTD2 mutations have not been described previously in AML.
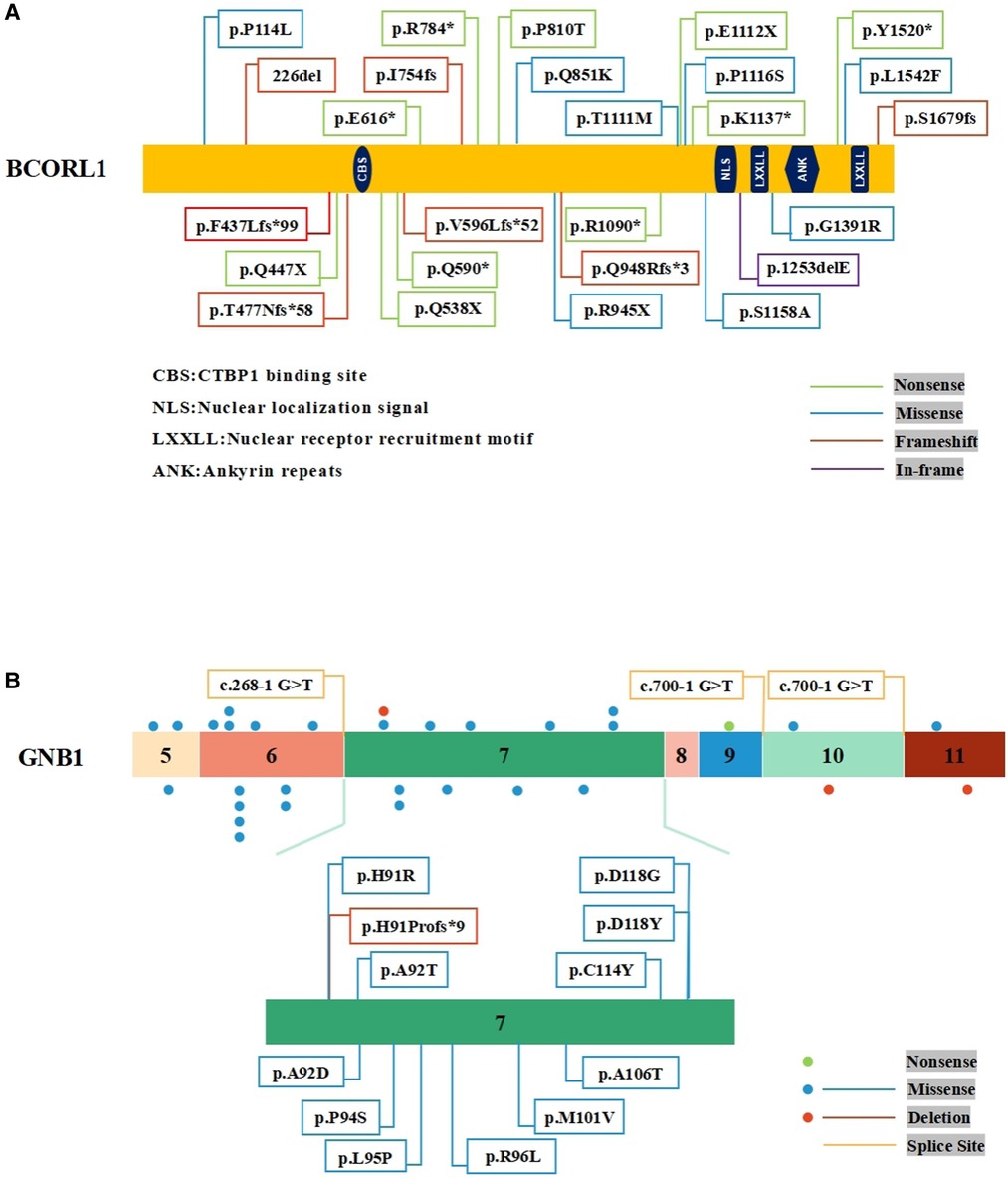
Figure 2. Schematic representation of BCORL1 and GNB1 mutations. (A) The mutations of BCORL1 in AML. (B) The mutations in Exon 7 of the GNB1 gene.
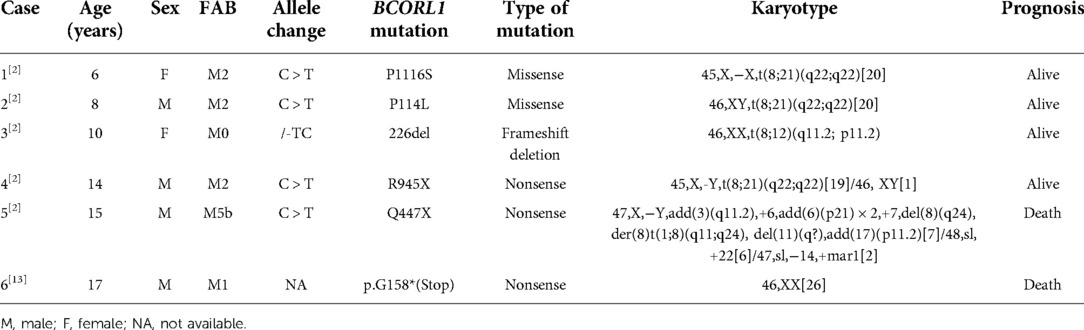
Table 2. Characteristics of pediatric AML with gene mutations in BCORL1.
AML is a heterogeneous disease characterized by the presence of different collaborating cytogenetic and molecular aberrations. Cytogenetic analysis had implicated several recurrent chromosomal structural abnormalities for pediatric AML pathogenesis, including 11q23 (KMT2A) rearrangements (frequency: 20%), t(8;21) (q22; q22) (frequency: 10%–12%), inv (16) (p13.1q22) or t(16;16) (p13.1;q22) (frequency: 10%), t(8;16) (p11;p13) (frequency:10%), t(15;17) (q24.1;q21.2) (frequency: 5%–10%), and t(6;9) (p22;q34) (frequency:1.2%–4%) (3). These acquired genetic abnormalities played an essential role in the pathogenesis of AML. Here, we described a pediatric AML with 48, XY, +13, +19[12]/48, idem, del(p12)[8]. In addition, sequencing technologies detected various mutations, including BCORL1, GNB1, SH2B3, and KMT2D. Although some of these mutations have rarely been reported in AML previously, mutations in the same region have been detected in patients with malignancies.
BCORL1 (S878G) somatic mutation has been found in intracranial germ cell tumors (16), but BCORL1 (S878G) germline mutation was reported in AML for the first time. To date, 26 different types of BCORL1 mutations have been reported and characterized among 572 adult AML patients, which included cases with secondary leukemia and 287 pediatric patients, including 10 nonsense mutations, 8 missense mutations, 7 frameshift mutations, and 1 in-frame mutation (Figure 2A). To a certain extent, these mutations may truncate the encoded proteins. Further, BCORL1 mutations were detected in the AML-193, SKM1, and OCI-AML5 cell lines, and all BCORL1 mutations in cell lines truncated the encoded protein as a result of nonsense mutations, splice site mutations, or out-of-frame insertions or deletions (10). Although not resulting in clearly diminished BCORL1 mRNA expression levels, these disruptive events were predicted to encode truncated proteins lacking the last C-terminal LXXLL nuclear receptor recruitment motif. This result not only implicates BCORL1 mutations in the pathogenesis of AML but also demonstrates that BCORL1 is a tumor suppressor gene inactivated by these mutations.
Mutations involving BCORL1 and other mutations were associated with adverse outcomes in AML. One pediatric AML had a normal karyotype but carried BCORL1 mutation [p.G158*(Stop)] and RUNX1 mutation and eventually died (13). Terada et al. reported that BCORL1 mutations had a significant positive correlation with RUNX1 mutations, which were thought to indicate an unfavorable prognosis in AML (11). In fact, BCORL1-mutated AML had concomitant other mutations, including KRAS, NRAS, IDH1 and IDH2, DNMT3A, RUNX1, and FLT3-ITD, and were mutually exclusive with TP53, CEBPA, or NPM1 mutations (10). However, due to the low number of co-mutations in BCORL1-mutated AML, additional studies are necessary to confirm prognostic information.
Mutations in GNB1 have been associated with myeloid and B-cell malignancies. Yoda et al. reported that GNB1 K57 mutations were in myeloid neoplasms, while most of the GNB1 I80 mutations were in B-cell neoplasms (17). GNB1 (G116S) somatic mutation has been reported in acute lymphoblastic B-cell leukemia (18), but GNB1 (G116S) mutation has not been described in AML. Moreover, an activating mutation of GNB1 (p.K89M) in ETV6-ABL1-positive leukemic cells led to a poor response to tyrosine kinase inhibitors therapy (19). These results suggest that GNB1 mutations may be closely related to hematological tumorigenesis. According to the published results, nearly 30 GNB1 mutation carriers have been identified so far. Most of the GNB1 mutations were located in Exon 6 and Exon 7, indicating the presence of a mutational hotspot. There have been found 12 mutations in Exon 7 of GNB1 (5, 20) (Figure 2B). Functional studies demonstrated that specific mutations affected the GNB1 residues that mediated interactions with both G proteins subunits and effector proteins. For example, the GNB1 mutation (p.M101V) affects a Gβ residue found important for (1) activation of adenylyl cyclase 2, (2) inhibition of calcium channels, (3) activation of phospholipase C-β2, and (4) G-protein-coupled receptors (21). In addition, GNB1 mutations activated AKT/mTOR signaling in human leukemia cells, and treatment with PI3K/mTOR inhibitor (BEZ235) suppressed PI3K-mTOR signaling and improved the survival of mice with mutant GNB1-induced leukemia (17). AML patients harbored leukemia cells that displayed dysregulation of the PI3K-Akt-mTOR signaling, and activation of this pathway also had a prognostic impact (22). Upregulated and dysregulated cytokine signaling is a major mechanism in the pathogenesis of multiple hematopoietic malignancies. Although the relevance remains unclear, we cautiously speculate that this paradigm might be applicable to GNB1-related AML.
The SH2B3 gene was found on chromosome 12q24.12, and the majority of SH2B3 mutations was missense mutations (23). Several SH2B3 mutations were identified in patients with MPNs or the related idiopathic erythrocytosis (W364X, S370C, L390W, R392Q, S394C, E395K, E400K, V402M, R415H, R415C, R425C, M437I, and I446V) and in patients with B-cell precursor ALL (R392W, R397G, and Q427P) (24). The mutational loss of function of SH2B3 in hematopoietic cells released its inhibitory activity against the activated tyrosine kinase receptors and its downstream JAK-STAT pathway, resulting in the enhanced proliferative capacity of hematopoietic cells. Wu et al. reported that SH2B3 (A536T) mutation was found in chronic myeloid leukemia (25). SH2B3 (A536T) mutation led to the substitution of alanine for threonine at position 536, which has not been reported in AML. The mechanisms of SH2B3 mutations in AML are unknown, and the potential effects require further investigation.
The KMT2D gene was mutated and deleted in many different types of cancer, including leukemia, bladder, lung, liver, prostate, breast, ovarian, gastric, pancreatic, renal, and colorectal cancers (26). Considering the effect of KMT2D on gene expression regulation, as well as its non-enzyme-dependent function, mutations of KMT2D may contribute to aberrant gene expression programs, thereby driving tumor malignancies. KMT2D had two frequently occurring genomic alterations: nonsense and frameshift mutations, which may generate truncated KMT2D proteins with a deficient methyltransferase activity (7, 27). Moreover, Kantidakis et al. pointed out that KMT2D mutation resulted in transcription stress and genome instability as a strong driver in tumorigenesis (28). However, KMTD2 (S3708R) mutation has not been described previously, and the significance and role of KMT2D mutations were not determined in AML.
To the best of our knowledge, this is the first reported case of pediatric AML with rare BCORL1, GNB1, SH2B3, and KMT2D mutations. This case highlights the possibility of an increased risk of hematological malignancies in individuals with co-occurring mutations, but the hierarchy and mechanism are poorly understood. Compared with the general population, this is an interesting study due to the rarity of this form and the resulting difficulty in finding patients to conduct further studies on. This paper aims to provide an opportunity to broaden the understanding available of this rare form of AML.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by the Medical Ethics Committee of Tianjin Children's Hospital, Tianjin Children's Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
LW and PS designed the study. SC performed the experiments. LW and YS collected samples and analyzed the results. LW wrote this article. PS revised it. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Creutzig U, Kutny MA, Barr R, Schlenk RF, Ribeiro RC. Acute myelogenous leukemia in adolescents and young adults. Pediatr Blood Cancer. (2018) 65:e27089. doi: 10.1002/pbc.27089
2. Shiba N, Yoshida K, Shiraishi Y, Okuno Y, Yamato G, Hara Y, et al. Whole-exome sequencing reveals the spectrum of gene mutations and the clonal evolution patterns in paediatric acute myeloid leukaemia. Br J Haematol. (2016) 175:476–89. doi: 10.1111/bjh.14247
3. Lejman M, Dziatkiewicz I, Jurek M. Straight to the point-the novel strategies to cure pediatric AML. Int J Mol Sci. (2022) 23:1968. doi: 10.3390/ijms23041968
4. Sportoletti P, Sorcini D, Falini B. BCOR gene alterations in hematologic diseases. Blood. (2021) 138:2455–68. doi: 10.1182/blood.2021010958
5. Da Silva JD, Costa MD, Almeida B, Lopes F, Maciel P, Teixeira-Castro A. Case report: a novel GNB1 mutation causes global developmental delay with intellectual disability and behavioral disorders. Front Neurol. (2021) 12:735549. doi: 10.3389/fneur.2021.735549
6. Maslah N, Cassinat B, Verger E, Kiladjian JJ, Velazquez L. The role of LNK/SH2B3 genetic alterations in myeloproliferative neoplasms and other hematological disorders. Leukemia. (2017) 31(8):1661–70. doi: 10.1038/leu.2017.139
7. Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med. (2015) 21:1190–8. doi: 10.1038/nm.3940
8. Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. (2017) 19(1):4–23. doi: 10.1016/j.jmoldx.2016.10.002
9. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
10. Li M, Collins R, Jiao Y, Ouillette P, Bixby D, Erba H, et al. Somatic mutations in the transcriptional corepressor gene BCORL1 in adult acute myelogenous leukemia. Blood. (2011) 118:5914–7. doi: 10.1182/blood-2011-05-356204
11. Terada K, Yamaguchi H, Ueki T, Usuki K, Kobayashi Y, Tajika K, et al. Usefulness of BCOR gene mutation as a prognostic factor in acute myeloid leukemia with intermediate cytogenetic prognosis. Genes Chromosomes Cancer. (2018) 57:401–8. doi: 10.1002/gcc.22542
12. Damm F, Chesnais V, Nagata Y, Yoshida K, Scourzic L, Okuno Y, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. (2013) 122:3169–77. doi: 10.1182/blood-2012-11-469619
13. de Rooij JD, van den Heuvel-Eibrink MM, Hermkens MC, Verboon LJ, Arentsen-Peters ST, Fornerod M, et al. BCOR and BCORL1 mutations in pediatric acute myeloid leukemia. Haematologica. (2015) 100:e194–5. doi: 10.3324/haematol.2014.117796
14. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. (2016) 374:2209–21. doi: 10.1056/NEJMoa1516192
15. Walter MJ, Shen D, Shao J, Ding L, White BS, Kandoth C, et al. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia. (2013) 27:1275–82. doi: 10.1038/leu.2013.58
16. Wang L, Yamaguchi S, Burstein MD, Terashima K, Chang K, Ng HK, et al. Novel somatic and germline mutations in intracranial germ cell tumours. Nature. (2014) 511(7508):241–5. doi: 10.1038/nature13296
17. Yoda A, Adelmant G, Tamburini J, Chapuy B, Shindoh N, Yoda Y, et al. Mutations in G protein β subunits promote transformation and kinase inhibitor resistance. Nat Med. (2015) 21:71–5. doi: 10.1038/nm.3751
18. Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. (2014) 371(11):1005–15. doi: 10.1056/NEJMoa1403088
19. Zimmermannova O, Doktorova E, Stuchly J, Kanderova V, Kuzilkova D, Strnad H, et al. An activating mutation of GNB1 is associated with resistance to tyrosine kinase inhibitors in ETV6-ABL1-positive leukemia. Oncogene. (2017) 36:5985–94. doi: 10.1038/onc.2017.210
20. Lohmann K, Masuho I, Patil DN, Baumann H, Hebert E, Steinrücke S, et al. Novel GNB1 mutations disrupt assembly and function of G protein heterotrimers and cause global developmental delay in humans. Hum Mol Genet. (2017) 26:1078–86. doi: 10.1093/hmg/ddx018
21. Petrovski S, Küry S, Myers CT, Anyane-Yeboa K, Cogné B, Bialer M, et al. Germline de novo mutations in GNB1 cause severe neurodevelopmental disability, hypotonia, and seizures. Am J Hum Genet. (2016) 98:1001–10. doi: 10.1016/j.ajhg.2016.03.011
22. Nepstad I, Hatfield KJ, Grønningsæter IS, Reikvam H. The PI3K-Akt-mTOR signaling pathway in human acute myeloid leukemia (AML) cells. Int J Mol Sci. (2020) 21:2907. doi: 10.3390/ijms21082907
23. Maslah N, Cassinat B, Verger E, Kiladjian JJ, Velazquez L. The role of LNK/SH2B3 genetic alterations in myeloproliferative neoplasms and other hematological disorders. Leukemia. (2017) 31:1661–70. doi: 10.1038/leu.2017.139
24. Morris R, Butler L, Perkins A, Kershaw NJ, Babon JJ. The role of LNK (SH2B3) in the regulation of JAK-STAT signalling in haematopoiesis. Pharmaceuticals (Basel). (2021) 15:24. doi: 10.3390/ph15010024
25. Wu W, Xu N, Zhou X, Liu L, Tan Y, Luo J, et al. Integrative genomic analysis reveals cancer-associated gene mutations in chronic myeloid leukemia patients with resistance or intolerance to tyrosine kinase inhibitor. Onco Targets Ther. (2020) 13:8581–91. doi: 10.2147/OTT.S257661
26. Wang LH, Aberin MAE, Wu S, Wang SP. The MLL3/4 H3K4 methyltransferase complex in establishing an active enhancer landscape. Biochem Soc Trans. (2021) 49:1041–54. doi: 10.1042/BST20191164
27. Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. (2015) 21:1199–208. doi: 10.1038/nm.3943
Keywords: acute myeloid leukemia, BCORL1, GNB1, SH2B3, KMT2D
Citation: Wang L, Chen S, Shen Y and Si P (2022) BCORL1 S878G, GNB1 G116S, SH2B3 A536T, and KMT2D S3708R tetramutation co-contribute to a pediatric acute myeloid leukemia: Case report and literature review. Front. Pediatr. 10:993952. doi: 10.3389/fped.2022.993952
Received: 14 July 2022; Accepted: 21 September 2022;
Published: 17 October 2022.
Edited by:
Julia Skokowa, University of Tübingen, GermanyReviewed by:
Eugenia Granado, National Cancer Institute (INCA), Brazil© 2022 Wang, Chen, Shen and Si. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Si a2FuZ3NpcGluZ0BsaXZlLmNu
Specialty Section: This article was submitted to Pediatric Hematology and Hematological Malignancies, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.