 Qi Huang1
Qi Huang1 Cheng Jiang
Cheng Jiang Jiazhong Sun
Jiazhong Sun Victor Wei Zhang
Victor Wei Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 18 November 2022
Sec. Genetics of Common and Rare Diseases
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.990230
This article is part of the Research TopicAdvancing Genomics for Rare Disease Diagnosis and Therapy Development Vol IIView all 42 articles
Tricho-rhino-phalangeal syndrome (TRPS) is a rare autosomal dominant malformation caused by mutations involving the TRPS1 gene. Patients with TRPS exhibit distinctive craniofacial and skeletal abnormalities. This report presents three intra-familial cases with TRPS1 gene mutations that showed the characteristic features of TRPS. A 13-year-old boy was admitted to Department of Endocrinology for the evaluation of short stature. Physical examination revealed that the boy had thin sparse hair, pear-shaped nose, protruding ears, small jaw and brachydactyly. A survey of his family history indicated that the boy's sister and mother shared the same clinical features. Radiological techniques demonstrated a different degree of skeletal abnormalities in these siblings. Next-generation sequencing and quantitative PCR were performed and showed a novel deletion mutation in exons 3–5 in the three familial cases, confirming the diagnosis of TRPS I. The healthy father did not carry the deletion mutation. Currently, there was no specific therapy for TRPS I; however, genetic consultation may be useful for family planning
First reported by Giedion in 1966 (1), Tricho-rhino-phalangeal syndrome (TRPS) is a rare heritable congenital or sporadic disorder characterized by typical craniofacial features and noticeable skeletal abnormalities, especially of phalanges, metacarpals and metatarsal bones (2–4).
Based on clinical characteristics and genetic analysis, TRPS is distinguished into three subtypes: TRPS I (OMIM 190350), known as Giedion syndrome, have distinct clinical manifestations that often correspond to distinct mutations or haploinsufficiency in the TRPS1 gene (5). Moreover, TRPS II (OMIM 150230), also named Langer-Giedion syndrome (LGS), is caused by a contiguous gene deletions involving both TRPS1 and EXT1 (6, 7). TRPS II differs from TRPS I by the presence of multiple exostoses and intellectual disability (6). TRPS III (OMIM 190351) is also associated with TRPS1 mutations. Besides typical TRPS feature, TRPS III cases have more severe skeletal malformations (7).
Herein, we describe a Chinese Han family with three TRPS I cases caused by a novel deletion mutation in the TRPS1 gene involving exons 3–5 (Figure 1).
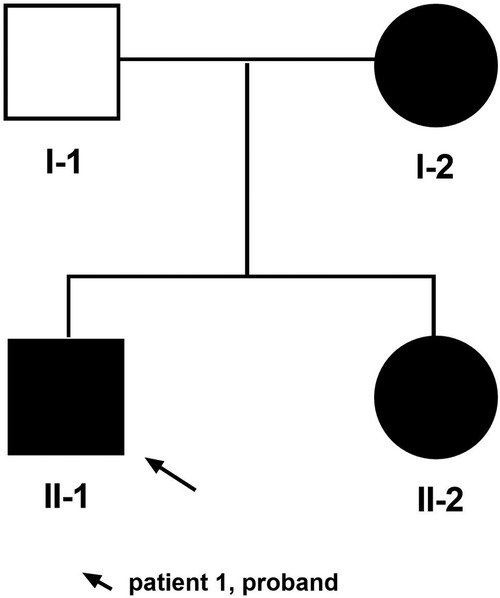
Figure 1. Pedigree of family. The arrow indicates patient 1 as the proband.
A 13-year-old boy, proband, was first admitted to our Endocrinology Department for evaluation of his short stature. He was born after a full term pregnancy and normal delivery, as the second child in non-consanguineous family. His parents reported that his birth weight and length was normal but gradually developed short stature upon birth. In addition, he often suffered from respiratory infections and his tonsils were removed. However, no intellectual impairment was observed.
Upon admission, a routine examination revealed that the boy's weight was 81 kg (>97th percentile) and his standing height was 152.2 cm (3rd percentile). Pubertal development was normal. Another prominent dysmorphic feature included markedly thin and sparse scalp hair, protruding ears, a bulbous pear-shaped nose and a long philtrum with a thin upper lip [Figure 2A(a)].
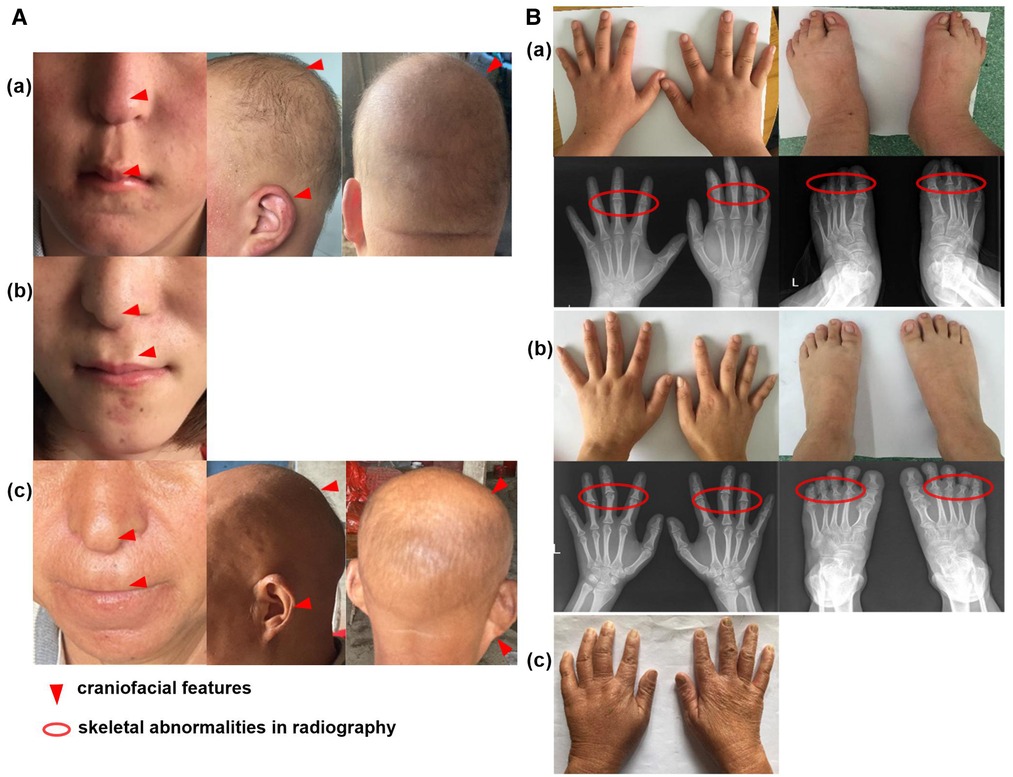
Figure 2. Clinical feature of the family cases. (A) The craniofacial feature of proband (a), his sister with wig (b) and mother (c); (B) The skeletal abnormalities of proband (a), his sister (b) and mother (c).
Laboratory tests showed that serum levels of calcium, inorganic phosphate, alkaline phosphatase, free T4, TSH, PTH, corticosteroid and insulin-like growth factor 1 (IGF-1) were normal. The karyotype was 46, XY. Further extremity and radiological examinations showed brachydactyly of fingers and toes [Figure 2B(a)].
The patient was a 23-year-old girl, the elder sister of patient 1. She showed similar features to her brother, with a height of 146 cm (<3rd percentile). She was almost bald and declined to take off her wig [Figure 2A(b)]. She showed a brachydactyly with obvious clinodactyly, a deviation of the forefinger, middle fingers and ring fingers bilaterally. Radiography revealed distortion of the proximal middle phalanges on the second, third and fourth fingers bilaterally. She also showed a skeletal malformation on second through fifth proximal phalanx on both feet [Figure 2B(b)].
The mother of the siblings, with a height of 140 cm (<3rd percentile), presented with sparse scalp hair and a nose with a bulbous tip [Figure 2A(c)]. She also showed bone abnormalities on her hands [Figure 2B(c)], but she declined further laboratory tests and radiological examinations.
Genomic DNA from the proband and his family members was extracted from peripheral blood samples. A custom-designed Medical Exome Sequencing (MES, AmCare Genomic Lab), including target region capture of more than 5,000 phenotype-related genes contained in the Online Mendelian Inheritance in Man (OMIM), was applied and was followed by next-generation sequencing (NGS, PE 150) on the Illumina platform (Illumina, Inc.). Alignment of the sequence to the reference human genome (hg19) was performed by NextGen (Softgenetics, LLC). Trio analysis including both SNV annotation and exome-based CNV identification was done by an in-house pipeline. Synonymous as well as common SNPs (MAF > 0.1% in gnomAD) were filtered out subsequently. All the candidate variants were further evaluated based on the ACMG guideline for SNV interpretation (8). A detailed protocol was described in a previous study (9).
The Candidate variant was validated by quantitative PCR (qPCR). Three pair primers of TRPS1 exon 3–5 were designed to amplify all exons of the deletion fragment as follows: TRPS1-EX3, 5′-TGAAACTGGGCTCAAACCTT-3′ (forward) and 5′-GGGG ACTCACTGGAGACAAA-3′ (reverse); TRPS1-EX4, 5′-CTGGTGGCCTCTGTACC ATT-3′ (forward) and 5′-ACAAAATA AAAGCTTCTCTCCCC-3′ (reverse); TRPS1-EX5, 5′- AGGAATCCCTTGGTTTCCAC -3′ (forward) and 5′-AGTCCGTCATACAC CCAAGC-3′ (reverse).
Based on the MES trio analysis and qPCR validation, a small heterozygous deletion c.38−? _2700+? del within the TRPS1 gene (NM_014112.5) was identified (Figure 3A). It is segregated in all the patients of this family (proband, his elder sister and mother), and the healthy father did not carry the deletion (Figures 3B,C). This novel small deletion includes exons 3 to 5, and is not present in gnomAD database, HGMD or any peer-reviewed publication. It is predicted to disrupt the reading frame and undergo nonsense-mediated decay (NMD) resulting in an amino acid change (p.Asn13Lysfs*3) because of the multiple exons deletion. According to the ACMG guideline, this variant is classified as likely pathogenic.
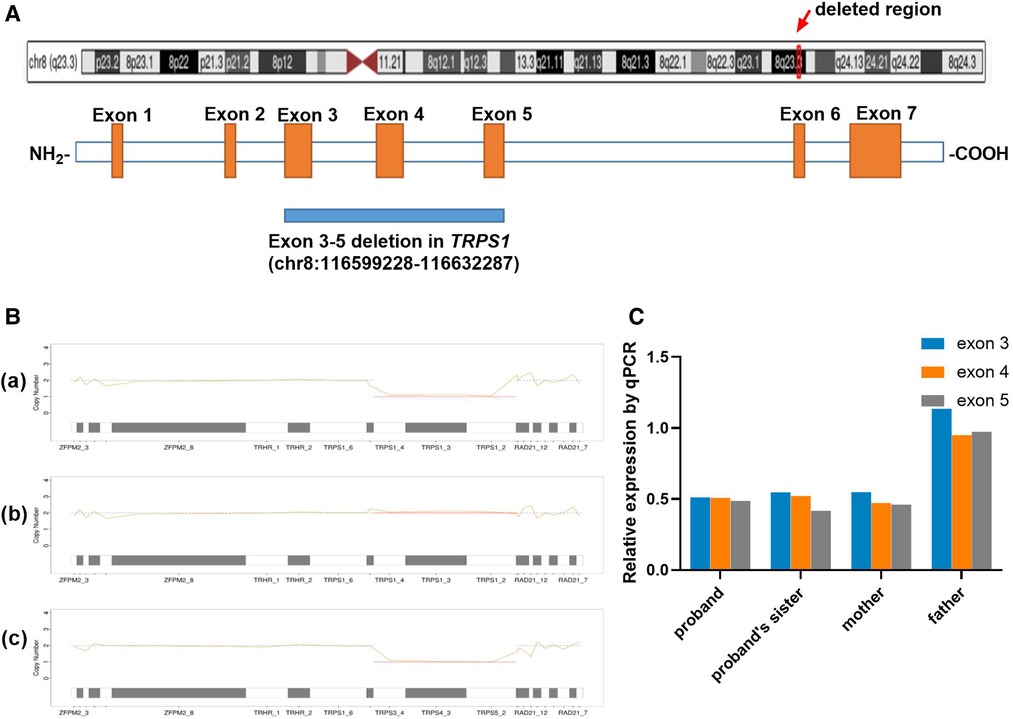
Figure 3. Genetic analysis. (A) Distribution of the muti-exon deletion in TRPS1 gene; (B) Copy number variation result of proband (a), his father (b) and mother (c) using NGS coverage depth data. The line charts present the copy number of 8q23 region for each family member, including TRPS1 and the gene's upstream and downstream locations. The proband and his mother were heterozygous for the deletion (×1) for exons 3–5 (CDS2-4), and his father was normal at this region. (C) qPCR results of all the family members. The proband, his sister and mother were carrying heterozygous deletion of exons 3–5 (×0.5), and his father was normal (×1).
TRPS1 was reported as the causal gene of TRPS I by Momeni et al. in 2000 (3). Haploinsufficiency is the known pathogenicity mechanism for the TRPS1 gene (7, 10). In a previous comprehensive study, deletion variants of TRPS1 have been reported in multiple cases, most of them are whole gene deletions that include exons 1 to 7 or large fragments deletions. Only one patient carrying a smaller (exon 2–6) deletion within the gene has been reported (2). The recurrence of variable sizes of fragment deletion suggests the structure complexity in this region.
We are reporting the second family carrying a small 3-exon deletion within the TRPS1 gene, which is predicted to disrupt the functional GATA motif of TRPS1. A mouse model study has revealed that a heterozygous knockdown of the GATA motif leads to hair and facial anomalies that overlap with findings of TRPS (11).
Our study also provides further evidence that structure variation is a common cause of TRPS. In this study, we used an optimized pipeline that combined both the SNV identification and NGS coverage depth data for CNV (even the small deletion/duplications) calls within one dataset, which proved to be a sensitive and cost-effective genetic analysis for the suspected TRPS patients, as well as for the better understanding of the genetic etiology of TRPS.
Definitive diagnosis of the disease is essential to perform timely therapeutic procedures. Nevertheless some alternative approaches have been tried for therapy of a few TRPS cases with mixed results. Short stature is a frequent clinical finding in affected individuals. How to improve their short stature is what these patients and their parents are most interested in. K Stagi (12) and Sarafoglou (13) described their TRPS I cases with or without growth hormone (GH) deficiency, and a remarkable increase in growth was observed through GH therapy in four cases. However, Naselli (14) reported another two TRPS I cases with poor growth, and showed no improvement in linear growth after a 1-year GH replacement therapy. In our study, the evaluation of GH-IGF-1 axis revealed that the boy did not have GH deficiency. His bone age was 15-year assessed through RUS-CHN radiographic atlas method, therefore he had no indications for GH treatment.
Sparse scalp hair is another major feature of TRPS patients. Their diffuse alopecia varies from normal hair to complete baldness (15), and the treatment option for alopecia remains unclear. Mi Soo Choi reported their experience in the medical treatment of a TRPS boy (15). In this case, neither topical minoxidil nor oral finasteride was effective in preventing the progression of alopecia or inducing hair growth. Finally, the patient's hairs started to re-grow at 4 months after hair transplantation operation. All of our three patients complained of hair loss and slow hair growth rate since their childhood. Compared to Mi Soo Choi's patient (15), whose occipital scalp hair had normal hair density and diameter, our patients' hair on the entire scalp was affected and tended to be thinner.
At present there is no special therapy for TRPS, even though alternative approaches were employed as GH replacement therapy for short stature and hair transplantation for baldness, the therapeutic results mixed. Therefore, genetic counseling may be useful for family planning.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Ethics committee of Zhongnan Hospital of Wuhan University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
QH: conducted research. QH and CJ: wrote the paper. QH, JZS and JLX: conceived the research. VWZ: Writing-analyzed the data. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the Natural Science Foundation of Hubei Province (2017CFB265) and National Natural Science Foundation of China (81800764).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Giedion A. Tricho-rhino-phalangeal syndrome. Helv Paediatr Acta. (1966) 21(5):475–85. Das tricho-rhino-phalangeale Syndrom. ger.5991804
2. Maas SM, Shaw AC, Bikker H, Lüdecke HJ, van der Tuin K, Badura-Stronka M, et al. Phenotype and genotype in 103 patients with tricho-rhino-phalangeal syndrome. Eur J Med Genet. (2015) 58(5):279–92. doi: 10.1016/j.ejmg.2015.03.002
3. Momeni P, Glöckner G, Schmidt O, von Holtum D, Albrecht B, Gillessen-Kaesbach G, et al. Mutations in a new gene, encoding a zinc-finger protein, cause tricho-rhino-phalangeal syndrome type I. Nat Genet. (2000) 24(1):71–4. doi: 10.1038/71717
4. Wang C, Xu Y, Qing Y, Yao R, Li N, Wang X, et al. TRPS1 Mutation detection in Chinese patients with tricho-rhino-phalangeal syndrome and identification of four novel mutations. Mol Genet Genomic Med. (2020) 8(10):e1417. doi: 10.1002/mgg3.1417
5. Kaiser FJ, Brega P, Raff ML, Byers PH, Gallati S, Kay TT, et al. Novel missense mutations in the TRPS1 transcription factor define the nuclear localization signal. Eur J Hum Genet. (2004) 12(2):121–6. doi: 10.1038/sj.ejhg.5201094
6. Shanske AL, Patel A, Saukam S, Levy B, Lüdecke HJ. Clinical and molecular characterization of a patient with langer-giedion syndrome and mosaic del(8)(q22.3q24.13). Am J Med Genet, Part A. (2008) 146a(24):3211–6. doi: 10.1002/ajmg.a.32615
7. Lüdecke HJ, Schaper J, Meinecke P, Momeni P, Gross S, von Holtum D, et al. Genotypic and phenotypic spectrum in tricho-rhino-phalangeal syndrome types I and III. Am J Hum Genet. (2001) 68(1):81–91. doi: 10.1086/316926
8. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
9. Wang Z, Lin J, Qiao K, Cai S, Zhang VW, Zhao C, et al. Novel mutations in HINT1 gene cause the autosomal recessive axonal neuropathy with neuromyotonia. Eur J Med Genet. (2019) 62(3):190–4. doi: 10.1016/j.ejmg.2018.07.009
10. Zepeda-Mendoza CJ, Cousin MA, Basu S, Jenkinson G, Oliver G, Pittock ST, et al. An intragenic duplication of TRPS1 leading to abnormal transcripts and causing trichorhinophalangeal syndrome type I. Cold Spring Harbor Mol Case Stud. (2019) 5(6):a004655. doi: 10.1101/mcs.a004655
11. Malik TH, Von Stechow D, Bronson RT, Shivdasani RA. Deletion of the GATA domain of TRPS1 causes an absence of facial hair and provides new insights into the bone disorder in inherited tricho-rhino-phalangeal syndromes. Mol Cell Biol. (2002) 22(24):8592–600. doi: 10.1128/MCB.22.24.8592-8600.2002
12. Stagi S, Bindi G, Galluzzi F, Lapi E, Salti R, Chiarelli F. Partial growth hormone deficiency and changed bone quality and mass in type I trichorhinophalangeal syndrome. Am J Med Genet, Part A. (2008) 146a(12):1598–604. doi: 10.1002/ajmg.a.32348
13. Sarafoglou K, Moassesfar S, Miller BS. Improved growth and bone mineral density in type I trichorhinophalangeal syndrome in response to growth hormone therapy. Clin Genet. (2010) 78(6):591–3. doi: 10.1111/j.1399-0004.2010.01434.x
14. Naselli A, Vignolo M, Di Battista E, Papale V, Aicardi G, Becchetti S, et al. Trichorhinophalangeal syndrome type I in monozygotic twins discordant for hip pathology. Report on the morphological evolution of cone-shaped epiphyses and the unusual pattern of skeletal maturation. Pediatr Radiol. (1998) 28(11):851–5. doi: 10.1007/s002470050481
Keywords: tricho-Rhino-Phalangeal syndrome, TRPS1 gene, deletion mutation, short stature, sparse hair
Citation: Huang Q, Jiang C, Sun J, Xue J and Zhang V (2022) Case report: A novel mutation in TRPS1 identified in a Chinese family with tricho-rhino-phalangeal syndrome I: A therapeutic challenge. Front. Pediatr. 10:990230. doi: 10.3389/fped.2022.990230
Received: 9 July 2022; Accepted: 2 November 2022;
Published: 18 November 2022.
Edited by:
Ruth Roberts, ApconiX, United KingdomReviewed by:
Robert Lebel, Upstate Medical University, United States© 2022 Huang, Jiang, Sun, Xue and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiazhong Sun c3VuamlhemhvbmczMDBAMTYzLmNvbQ== Junli Xue bDEwMDQ1QHlhbmd0emV1LmVkdS5jbg==
Specialty Section: This article was submitted to Genetics of Common and Rare Diseases, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.