 Petra Loid1,2,3*
Petra Loid1,2,3* Marita Lipsanen-Nyman2,3Sirpa Ala-Mello4Katariina Hannula-Jouppi1,5,6
Marita Lipsanen-Nyman2,3Sirpa Ala-Mello4Katariina Hannula-Jouppi1,5,6 Juha Kere1,6,7
Juha Kere1,6,7 Outi Mäkitie1,2,3,8
Outi Mäkitie1,2,3,8 Mari Muurinen1,2,3
Mari Muurinen1,2,3
- 1Folkhälsan Research Center, Genetics Research Program, Helsinki, Finland
- 2Pediatric Research Center, Children's Hospital, University of Helsinki and Helsinki University Hospital, Helsinki, Finland
- 3Research Program for Clinical and Molecular Metabolism, University of Helsinki, Helsinki, Finland
- 4Department of Clinical Genetics, Helsinki University Hospital, Helsinki, Finland
- 5Department of Dermatology and Allergology, University of Helsinki and Helsinki University Hospital, Helsinki, Finland
- 6Stem Cells and Metabolism Research Program, University of Helsinki, Helsinki, Finland
- 7Department of Biosciences and Nutrition, Karolinska Institutet, Stockholm, Sweden
- 8Department of Molecular Medicine and Surgery, Karolinska Institutet, and Clinical Genetics, Karolinska University Hospital, Stockholm, Sweden
Silver-Russell syndrome (SRS, OMIM 180860) is a rare imprinting disorder characterized by intrauterine and postnatal growth restriction, feeding difficulties in early childhood, characteristic facial features, and body asymmetry. The molecular cause most commonly relates to hypomethylation of the imprinted 11p15.5 IGF2/H19 domain but remains unknown in about 40% of the patients. Recently, heterozygous paternally inherited pathogenic variants in IGF2, the gene encoding insulin-like growth factor 2 (IGF2), have been identified in patients with SRS. We report a novel de novo missense variant in IGF2 (c.122T > G, p.Leu41Arg) on the paternally derived allele in a 16-year-old boy with a clinical diagnosis of SRS. The missense variant was identified by targeted exome sequencing and predicted pathogenic by multiple in silico tools. It affects a highly conserved residue on a domain that is important for binding of other molecules. Our finding expands the spectrum of disease-causing variants in IGF2. Targeted exome sequencing is a useful diagnostic tool in patients with negative results of common diagnostic tests for SRS.
Introduction
Silver-Russell syndrome (SRS, OMIM 180860) is a rare imprinting disorder characterized by intrauterine and postnatal growth restriction, feeding difficulties and/or low body mass index in early childhood, relative macrocephaly at birth, prominent forehead in infancy and body asymmetry (1). Clinical diagnosis of SRS can be made in patients who have at least four of the six abovementioned clinical features according to the Netchine-Harbison Clinical Scoring System (NH-CSS) (2). The estimated incidence of SRS is 1/75 000–1/100 000 (3).
IGF2 is an imprinted gene expressed from the paternal allele, except in adult liver and central nervous system (4). IGF2 encodes insulin-like growth factor 2 (IGF2), a hormone important for fetal growth and development. Hypomethylation of the imprinted 11p15.5 IGF2/H19 domain leading to downregulation of IGF2 is the main molecular cause of SRS (seen in 30%–60% of patients). Maternal uniparental disomy of chromosome 7 [UPD(7)mat] is another common cause of SRS (seen in 5%–10% of patients) (1, 5). However, among patients with SRS the molecular cause remains unknown in about 40% of the patients (5). Next generation sequencing has been proposed as a useful tool to increase the diagnostic yield in SRS (6–8). Recently, rare pathogenic/likely pathogenic single nucleotide variants (SNVs) and/or copy number variants (CNVs) involving IGF2, CDKN1C, HMGA2, PLAG1 have been associated with SRS (6, 7, 9–18).
To date, only 16 pathogenic/likely pathogenic IGF2 variants have been reported in patients with SRS- like phenotype (6, 8, 11–18). In this study, we report a novel de novo missense variant in IGF2 identified by targeted exome sequencing in a Finnish patient with a clinical diagnosis of SRS. Our finding expands the spectrum of disease-causing variants in IGF2.
Methods
This study was carried out at Children's Hospital, Helsinki University Hospital and Folkhälsan Research Center in Helsinki, Finland. This study is a part of a research project investigating genetic causes of growth restriction and Silver-Russell syndrome. The inclusion criteria for the study were: at least −2.5 standard deviation score (SDS) pre- and/or postnatal growth restriction, normal chromosomes, and growth restriction of unknown etiology. Participants with an established molecular diagnosis were excluded from the study. Written informed consents were obtained from all participants or their parents/guardians. Ethical approval for this study was obtained from the research ethics committee of the Hospital District of Helsinki and Uusimaa. We used a custom-made targeted exome sequencing panel including 566 genes associated with growth and skeletal disorders. The original raw data consist of a clinical exome sequencing assay of more than 4,000 genes, but only selected genes were analyzed. The gene panel includes all protein coding exons and 20 base pairs from exon-intron boundary. In addition, the panel includes known disease-associated non-coding, deep intronic variants and regulatory variants. Panel testing included sequence and copy number variation analyses of the 566 genes in the panel. The targeted exome sequencing was performed at Blueprints Genetics, Helsinki, Finland. Sequence reads were aligned to reference human genome (GRCh 37/hg19). Burrows-Wheeler Aligner software was used for read alignment and variant calling were performed using GATK. CNV analysis was performed by Blueprint Genetics Ltd. (a Quest clinical laboratory), using an in-house developed pipeline, which has been validated in the CLIA (Clinical Laboratory Improvement Amendments) and CAP (College of American Pathologists) accredited Blueprint Genetics diagnostic laboratory. Only regions covered by the panel were included in the CNV analysis. We performed Sanger sequencing to confirm the findings and to analyze parental samples. The HOPE tool (https://www3.cmbi.umcn.nl/hope) was used to predict the structural effect of amino acid change on protein conformation.
Results
Clinical features
We report a 16-year-old boy born at 37 + 2 weeks of gestation to healthy parents of Finnish descent. Fetal growth failure was diagnosed by ultrasound at gestational week 19 and he was born very small for gestational age with birth length 38.5 cm (−6.1 SDS), birth weight 1480 g (−4.3 SDS) and a head circumference of 30.5 cm (−3.2 SDS). He had relative macrocephaly, with head circumference 2.9 SDS above birth length SDS. Weight of the placenta was 260 g, which is remarkably lower than the 3rd centile placental weight reported in a Scandinavian cohort (426 g for 261 gestational days, male sex and multiparity) (19). Figure 1 presents the growth charts of the patient. From the age of 3 months, he experienced spontaneous catch-up growth; the length SDS was −6.6 at 3 months, −5.5 at 6 months, and −3.4 at 3 years of age. Postnatally he suffered from severe feeding difficulties. Weight development with both nutritional and caloric supplement was good. He received gastric tube feeding from the age of 5 months and had a percutaneous endoscopic gastrostomy inserted at 8 months. He presented with a triangular-shaped face, small chin, thin lips, and 5th finger clinodactyly. He had no body asymmetry. Brain ultrasound and echocardiogram were normal. He had normal chromosomes, and genetic testing for GRACILE syndrome and Mulibrey nanism were normal. Silver-Russell Syndrome was suspected early neonatally. He was tested for UPD(7)mat and 11p15 methylation with normal results. A clinical diagnosis of SRS was made. He did not present with any cognitive, motor or speech delay. From the age of 3 years, he received recombinant human growth hormone (rhGH) treatment to increase adult height. Pretreatment investigations presented normal growth hormone (GH), high to normal serum insulin-like growth factor 1 (IGF1) and normal insulin-like growth factor-binding protein 3 (IGFBP3) levels. During GH treatment, serum IGF1 level was consistently at a high range, with normal to low GH doses. GH therapy accelerated the growth velocity; height SDS was −3.4 at the start of therapy, −2.8 after 1 year of treatment, −2.5 after 2 years of treatment, and −1.7 at pubertal onset. Pubertal timing was normal. Testicular size was small. In early childhood serum FSH, LH, testosterone and inhibinB were measured twice and the levels were normal. Testicular growth and virilization proceeded close to normal. At the end of the puberty, testicular size was in low normal range (testicular length 30 mm) and the secondary sex characteristics had reached stage P5G4. The attained final height at age 16 years was 160 cm (−2.2 SDS).
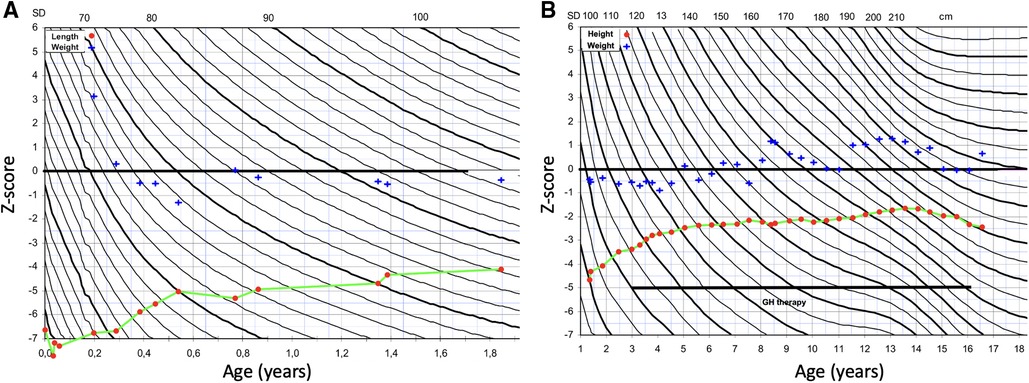
Figure 1. Growth charts of the patient. (A) Z-score for length (red dot) and weight-for-length (blue cross) at 0–2 years of age. (B) Z-score for height (red dot) and weight-for-height at 1–16 years of age. Growth hormone (GH) therapy was started at 3 years of age.
Genetic findings
The custom-made targeted exome sequencing included sequence and copy number variation analyses of 566 genes. Median read depth for the analysis was 234-fold, and 99.78% of target nucleotides were covered with >20-fold read depth. CNV analysis did not detect any known disease-causing or novel CNVs that were considered pathogenic. A novel de novo heterozygous missense variant NM_000612.5 (IGF2) c.122T > G, p.Leu41Arg was identified in the patient. The identified variant has not been observed in gnomAD, 1,000 Genomes, or Sequencing Initiative Suomi database (SISu), and has not been reported in dbSNP, ClinVar, or HGMD. This variant was predicted as pathogenic/damaging by SIFT, MutationTaster2, M-CAP, MVP, Provean, REVEL and PrimateAI. Polyphen2 predicted the variant as probably damaging. This variant has a CADD score 27.4.
The missense variant is located in the disulfide bond domain of IGF2 and affects a highly conserved residue (PhyloP 100-way score of 7.912). Analysis of the mutant protein by HOPE showed that the mutant residue is bigger, more hydrophilic and introduces a positive charge compared to the smaller, neutral charged wildtype residue (Figure 2).
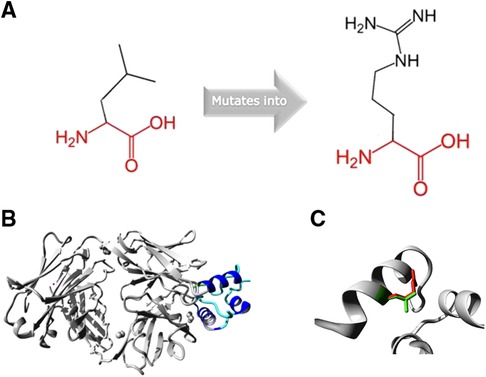
Figure 2. (A) Schematic structure of the original and the mutant amino acid. (B) Overview of the protein in ribbon-presentation. The protein is coloured by element; α-helix = blue and random coil = cyan. (C) Close-up of the mutation. The protein is coloured grey, the side chains of the wild-type (green) and the mutant residue (9).
The IGF2 missense variant was confirmed by Sanger sequencing and was absent in the parents (Figure 3A). We investigated the parental origin of the IGF2 missense variant by examining a common SNP (rs3213225) located 96 bp away from the variant. The child and father were heterozygous (G/A) for the SNP and the mother was homozygous (A/A) (Figure 3B). By exploring the reads in the Integrative Genomics Viewer (IGV), we found that the reads with SNP reference allele G also carried the missense variant indicating that the missense variant occurred on the paternal allele (Figure 3C).
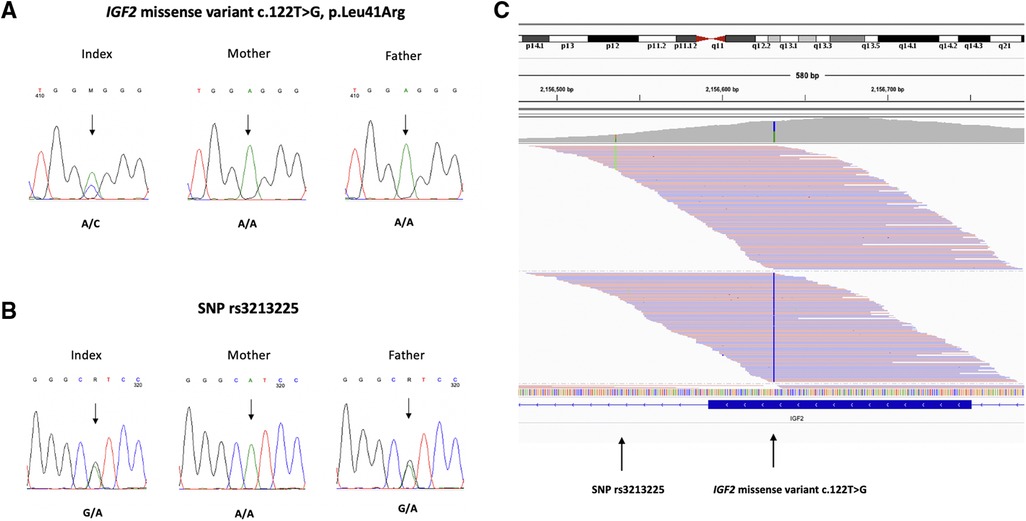
Figure 3. (A) Chromatograms of direct sequence analysis of IGF2 gene showing the missense variant c.122T > G, p.Leu41Arg in the patient (B) the SNP rs3213225 in the patient and father (C) IGV view of the patient's sequence data shows that the reads with SNP alternative allele A (in green) and the missense variant c.122T > G (in blue) are on different reads. The father is heterozygous (G/A) for the SNP and the mother is homozygous (A/A). The reads with the SNP reference allele G, that also carry the missense variant c.122T > G, must be paternal.
Table 1 presents the clinical features of our patient and the patients with previously reported IGF2 variants.

Table 1. Clinical characteristics in our patient and patients with previously reported IGF2 variants (6, 8, 11–18).
Discussion
SRS can be caused by several different genetic mechanisms including epimutations, uniparental disomy, rare pathogenic single nucleotide variants and copy number variants. About 40% of patients with SRS phenotype remain without a molecular diagnosis. Recently, next generation sequencing has been suggested as a useful tool to improve the identification of novel variants associated with SRS (6, 8). We describe a novel de novo IGF2 missense variant identified by targeted exome sequencing in a patient with a clinical diagnosis of SRS. The missense variant was predicted pathogenic by multiple in silico tools and affects a highly conserved residue. The variant was also confirmed to be on the paternal allele, consistent with the paternal expression pattern of IGF2.
To our knowledge, only 16 pathogenic/likely pathogenic IGF2 variants have been reported in 20 patients with SRS-like phenotype but the prevalence of IGF2 variants in large SRS populations remains unknown (6, 8, 11–18). The previously described IGF2 variants include eight missense, three splicing, three frameshift and two stop mutations. All patients presented with pre- and postnatal growth restriction and feeding difficulties. Our patient had severe prenatal growth restriction and feeding difficulties, but he had spontaneous catch-up growth from the age of 3 months. He had stable weight development though he required feeding measures. Postnatal catch-up growth is not usually seen in patients with SRS, including patients with IGF2 variants. 90% of the patients with IGF2 variants had relative macrocephaly, a prominent forehead, and a triangular face. These features were also observed in our patient. He did not present with any developmental delay or intellectual disability, which has been reported in 80% of the patients with IGF2 variants. However, in five out of twenty patients, there was no information about developmental delay/intellectual disability. Notably, our patient had high serum IGF1 level, which has also been reported in other patients with IGF2 variants (11, 14, 18). Masunga et al. compared clinical features and endocrine findings between patients with IGF2 variants and those with H19/IGF2:IG-DMR epimutations and found that IGF2 variants were associated with higher serum IGF1 level. They also reported that IGF2 variants were associated with high frequency of cardiovascular anomalies and developmental delay, and low frequency of body asymmetry compared to H19/IGF2:IG-DMR epimutations (14). Our patient had no body asymmetry, which may be one of the clinical features that best differentiates IGF2 variants from SRS caused by epigenetic changes.
Genetic counseling is recommended for patients with SRS and their families. The recurrence risk depends on the underlying cause of SRS and the sex of the transmitting parent. In most cases, only one child is affected and the recurrence risk for SRS in cases of de novo loss of paternal methylation of H19/IGF2 ICR1 or UPD(7)mat is very low. However, when SRS is caused by CNVs or pathogenic variants the recurrence risk can be up to 50%. In our case with a de novo pathogenic variant in IGF2, the recurrence risk is 50% for the offspring of the proband. Genetic testing is important for accurate genetic counseling.
IGF2 is an important prenatal growth factor and regulates the fetal demand and placental supply of nutrients (20). Small hypoplastic placentas and decreased levels of IGF2 have been found in patients with SRS (12, 14, 18). A hypoplastic placenta was also found in our patient. A recent mouse study revealed a direct role for the imprinted igf2-igf2r axis on matching placental development to fetal growth and showed that IGF2 produced by the fetus plays an important role in controlling placental microvasculature and trophoblast morphogenesis in late gestation (21). They suggest that poor placentation in fetal growth restriction may be due to deficient microvasculature expansion caused by decreased IGF2 signaling from the fetus. Another animal study proposed that growth anomalies in IGF2-dependent SRS can be detected prenatally by measuring IGF2 peptide levels in the amniotic fluid and prevented by prenatal genetic rescue targeting IGF2 (22). More studies are needed to investigate IGF2-based prenatal diagnosis and intervention strategies for IGF2-dependent SRS.
In conclusion, our finding expands the molecular and phenotypic spectrum of disease-causing variants in SRS and highlights the importance of screening for pathogenic variants in IGF2, especially when common diagnostic tests for SRS are negative. Next generation sequencing will likely increase the diagnostic yield of SRS. Whole exome and genome sequencing can identify novel genes associated with SRS or genes associated with differential diagnosis of SRS. Molecular diagnosis is required for specific genetic counseling and targeted clinical management.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Research ethics committee of the Hospital District of Helsinki and Uusimaa. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
Study design: MM, OM, ML-N, KH-J, JK. Data collection: all authors. Data analysis and data interpretation: all authors. Drafting manuscript: PL, MM, ML-N, OM. Revising manuscript content: All authors contributed to the article and approved the submitted version.
Funding
This study was supported by Sigrid Jusélius Foundation, Novo Nordisk Foundation, Academy of Finland, Folkhälsan Research Foundation and Foundation for Paediatric Research.
Acknowledgments
We thank the family for participation in this study. We thank Eira Leinonen, Mira Aronen and Ekateriina Pylsy for excellent assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Saal HM, Harbison MD, Netchine I, Adam MP, Everman DB, Mirzaa GM, et al. Silver-Russell syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, editors. Genereviews(®). Seattle (WA): University of Washington, Seattle; Copyright © 1993-2020, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle (2002). All rights reserved.
2. Azzi S, Salem J, Thibaud N, Chantot-Bastaraud S, Lieber E, Netchine I, et al. A prospective study validating a clinical scoring system and demonstrating phenotypical-genotypical correlations in Silver-Russell syndrome. J Med Genet. (2015) 52(7):446–53. doi: 10.1136/jmedgenet-2014-102979
3. Eggermann T, Davies JH, Tauber M, van den Akker E, Hokken-Koelega A, Johansson G, et al. Growth restriction and genomic imprinting-overlapping phenotypes support the concept of an imprinting network. Genes. (2021) 12(4):585. doi: 10.3390/genes12040585
4. Bergman D, Halje M, Nordin M, Engström W. Insulin-like growth factor 2 in development and disease: a mini-review. Gerontology. (2013) 59(3):240–9. doi: 10.1159/000343995
5. Wakeling EL, Brioude F, Lokulo-Sodipe O, O'Connell SM, Salem J, Bliek J, et al. Diagnosis and management of Silver-Russell syndrome: first international consensus statement. Nat Rev Endocrinol. (2017) 13(2):105–24. doi: 10.1038/nrendo.2016.138
6. Alhendi ASN, Lim D, McKee S, McEntagart M, Tatton-Brown K, Temple IK, et al. Whole-genome analysis as a diagnostic tool for patients referred for diagnosis of Silver-Russell syndrome: a real-world study. J Med Genet. (2022) 59(6):613–22. doi: 10.1136/jmedgenet-2021-107699
7. Inoue T, Nakamura A, Iwahashi-Odano M, Tanase-Nakao K, Matsubara K, Nishioka J, et al. Contribution of gene mutations to Silver-Russell syndrome phenotype: multigene sequencing analysis in 92 etiology-unknown patients. Clin Epigenetics. (2020) 12(1):86. doi: 10.1186/s13148-020-00865-x
8. Meyer R, Begemann M, Hübner CT, Dey D, Kuechler A, Elgizouli M, et al. One test for all: whole exome sequencing significantly improves the diagnostic yield in growth retarded patients referred for molecular testing for Silver-Russell syndrome. Orphanet J Rare Dis. (2021) 16(1):42. doi: 10.1186/s13023-021-01683-x
9. Fernández-Fructuoso JR, De la Torre-Sandoval C, Harbison MD, Chantot-Bastaraud S, Temple K, Lloreda-Garcia JM, et al. Silver russell syndrome in a preterm girl with 8q12.1 deletion encompassing PLAG1. Clin Dysmorphol. (2021) 30(4):194–6. doi: 10.1097/MCD.0000000000000375
10. Brereton RE, Nickerson SL, Woodward KJ, Edwards T, Sivamoorthy S, Ramos Vasques Walters F, et al. Further heterogeneity in Silver-Russell syndrome: pLAG1 deletion in association with a complex chromosomal rearrangement. Am J Med Genet A. (2021) 185(10):3136–45. doi: 10.1002/ajmg.a.62391
11. Abi Habib W, Brioude F, Edouard T, Bennett JT, Lienhardt-Roussie A, Tixier F, et al. Genetic disruption of the oncogenic HMGA2-PLAG1-IGF2 pathway causes fetal growth restriction. Genet Med. (2018) 20(2):250–8. doi: 10.1038/gim.2017.105
12. Begemann M, Zirn B, Santen G, Wirthgen E, Soellner L, Büttel HM, et al. Paternally inherited IGF2 mutation and growth restriction. N Engl J Med. (2015) 373(4):349–56. doi: 10.1056/NEJMoa1415227
13. Liu D, Wang Y, Yang XA, Liu D. De novo mutation of paternal IGF2 gene causing Silver-Russell syndrome in a sporadic patient. Front Genet. (2017) 8:105. doi: 10.3389/fgene.2017.00105
14. Masunaga Y, Inoue T, Yamoto K, Fujisawa Y, Sato Y, Kawashima-Sonoyama Y, et al. IGF2 mutations. J Clin Endocrinol Metab. (2020) 105(1):dgz034. doi: 10.1210/clinem/dgz034
15. Poulton C, Azmanov D, Atkinson V, Beilby J, Ewans L, Gration D, et al. Silver russel syndrome in an aboriginal patient from Australia. Am J Med Genet A. (2018) 176(12):2561–3. doi: 10.1002/ajmg.a.40502
16. Rockstroh D, Pfäffle H, Le Duc D, Rößler F, Schlensog-Schuster F, Heiker JT, et al. A new p.(Ile66Serfs*93) IGF2 variant is associated with pre- and postnatal growth retardation. Eur J Endocrinol. (2019) 180(1):K1–K13. doi: 10.1530/EJE-18-0601
17. Xia CL, Lyu Y, Li C, Li H, Zhang ZT, Yin SW, et al. Rare de novo IGF2 variant on the paternal allele in a patient with Silver-Russell syndrome. Front Genet. (2019) 10:1161. doi: 10.3389/fgene.2019.01161
18. Yamoto K, Saitsu H, Nakagawa N, Nakajima H, Hasegawa T, Fujisawa Y, et al. De novo IGF2 mutation on the paternal allele in a patient with Silver-Russell syndrome and ectrodactyly. Hum Mutat. (2017) 38(8):953–8. doi: 10.1002/humu.23253
19. Flatley C, Sole-Navais P, Vaudel M, Helgeland Ø, Modzelewska D, Johansson S, et al. Placental weight centiles adjusted for age, parity and fetal sex. Placenta. (2022) 117:87–94. doi: 10.1016/j.placenta.2021.10.011
20. Constância M, Hemberger M, Hughes J, Dean W, Ferguson-Smith A, Fundele R, et al. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature. (2002) 417(6892):945–8. doi: 10.1038/nature00819
21. Sandovici I, Georgopoulou A, Pérez-García V, Hufnagel A, López-Tello J, Lam BYH, et al. The imprinted Igf2-Igf2r axis is critical for matching placental microvasculature expansion to fetal growth. Dev Cell. (2022) 57(1):63.e8–79.e8. doi: 10.1016/j.devcel.2021.12.005
Keywords: IGF2, short stature, Silver-Russell, intrauterine growth restriction, exome sequencing
Citation: Loid P, Lipsanen-Nyman M, Ala-Mello S, Hannula-Jouppi K, Kere J, Mäkitie O and Muurinen M (2022) Case report: A novel de novo IGF2 missense variant in a Finnish patient with Silver-Russell syndrome. Front. Pediatr. 10:969881. doi: 10.3389/fped.2022.969881
Received: 10 August 2022; Accepted: 15 September 2022;
Published: 4 October 2022.
Edited by:
Katrin Õunap, University of Tartu, EstoniaReviewed by:
Deborah Mackay, University of Southampton, United KingdomThomas Eggermann, RWTH Aachen University, Germany
Frederic Brioude, Assistance Publique Hopitaux De Paris, France
© 2022 Loid, Lipsanen-Nyman, Ala-Mello, Hannula-Jouppi, Kere, Mäkitie and Muurinen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Petra Loid cGV0cmEubG9pZEBoZWxzaW5raS5maQ==
Specialty Section: This article was submitted to Pediatric Endocrinology, a section of the journal Frontiers in Pediatrics