
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 22 November 2022
Sec. Pediatric Hematology and Hematological Malignancies
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.967417
 Giuseppe Lassandro1*
Giuseppe Lassandro1* Valentina Palladino1
Valentina Palladino1 Michela Faleschini2
Michela Faleschini2 Angelica Barone3
Angelica Barone3 Gianluca Boscarol4
Gianluca Boscarol4 Simone Cesaro5Elena Chiocca6
Simone Cesaro5Elena Chiocca6 Piero Farruggia7Fiorina Giona8Chiara Gorio9Angela Maggio10Maddalena Marinoni11
Piero Farruggia7Fiorina Giona8Chiara Gorio9Angela Maggio10Maddalena Marinoni11 Antonio Marzollo12Giuseppe Palumbo13,14
Antonio Marzollo12Giuseppe Palumbo13,14 Giovanna Russo15
Giovanna Russo15 Paola Saracco16Marco Spinelli17
Paola Saracco16Marco Spinelli17 Federico Verzegnassi2
Federico Verzegnassi2 Francesca Morga1Anna Savoia2,18
Francesca Morga1Anna Savoia2,18 Paola Giordano1
Paola Giordano1
Background: Inherited thrombocytopenias (ITs) are rare congenital bleeding disorders characterized by different clinical expression and variable prognosis. ITs are poorly known by clinicians and often misdiagnosed with most common forms of thrombocytopenia.
Material and methods: “CHildren with Inherited Platelet disorders Surveillance” study (CHIPS) is a retrospective – prospective observational cohort study conducted between January 2003 and January 2022 in 17 centers affiliated to the Italian Association of Pediatric Hematology and Oncology (AIEOP). The primary objective of this study was to collect clinical and laboratory data on Italian pediatric patients with inherited thrombocytopenias. Secondary objectives were to calculate prevalence of ITs in Italian pediatric population and to assess frequency and genotype–phenotype correlation of different types of mutations in our study cohort.
Results: A total of 139 children, with ITs (82 male - 57 female) were enrolled. ITs prevalence in Italy ranged from 0.7 per 100,000 children during 2010 to 2 per 100,000 children during 2022. The median time between the onset of thrombocytopenia and the diagnosis of ITs was 1 years (range 0 - 18 years). A family history of thrombocytopenia has been reported in 90 patients (65%). Among 139 children with ITs, in 73 (53%) children almost one defective gene has been identified. In 61 patients a pathogenic mutation has been identified. Among them, 2 patients also carry a variant of uncertain significance (VUS), and 4 others harbour 2 VUS variants. VUS variants were identified in further 8 patients (6%), 4 of which carry more than one variant VUS. Three patients (2%) had a likely pathogenic variant while in 1 patient (1%) a variant was identified that was initially given an uncertain significance but was later classified as benign. In addition, in 17 patients the genetic diagnosis is not available, but their family history and clinical/laboratory features strongly suggest the presence of a specific genetic cause. In 49 children (35%) no genetic defect were identified. In ninetyseven patients (70%), thrombocytopenia was not associated with other clinically apparent disorders. However, 42 children (30%) had one or more additional clinical alterations.
Conclusion: Our study provides a descriptive collection of ITs in the pediatric Italian population.
Inherited thrombocytopenias (ITs) are a heterogeneous group of congenital bleeding disorders characterized by a reduced platelet count and variable clinical course. To date, a total of 45 different forms of ITs have been identified with different clinical expressions and variable prognosis. Main forms of ITs are exclusively characterized by a decreased platelet count with bleeding symptoms that vary in severity, ranging from severe clinical presentations, which may be revealed immediately after birth, to mild clinical presentations that could remain undiagnosed until fortuitous identification during routine laboratory examinations (1–3). Despite bleedings being considered the main clinical manifestation for patients with inherited thrombocytopenias, ITs are frequently associated with other congenital defects or an increased risk of developing further diseases such as hematological malignancies and kidney failure.
The prevalence of ITs in Europe is reported to be 2 per 1 million children (4). Although ITs are rare, latest advances in understanding these disorders suggested that their prevalence may be higher than previously thought (5, 6). Making a correct diagnosis of ITs may be difficult and often delayed because of the rarity of these conditions and their not specific clinical presentation. Moreover, ITs are often unrecognized and misdiagnosed with most common forms of thrombocytopenia. In addition, in more than of 50% of patients with ITs, the molecular cause remains unknown (7, 8).
To date, several studies describe the clinical picture and genetic characterization of ITs; they are usually focused on specific forms of ITs and report single-center case experiences (9, 10). However, larger further studies are needed to improve the clinical assessment and standardization of diagnosis of patients with ITs and to evaluate the efficacy of innovative therapeutic approaches (e.g., thrombopoietin receptor agonists) used successfully in acquired thrombocytopenias (11–13).
This multicenter retrospective–prospective study is the first one that provides a comprehensive overview of clinical, laboratory, and long-term outcomes of a large cohort of Italian children affected by ITs with the aim to improve knowledge and clinical management of these disorders.
The “CHildren with Inherited Platelet disorders Surveillance” study (CHIPS) is a retrospective–prospective observational cohort study conducted between January 2003 and January 2022 in 17 centers affiliated to the Italian Association of Pediatric Hematology and Oncology (AIEOP).
The primary objective of this study was to collect clinical and laboratory data on Italian pediatric patients with ITs. Secondary objectives were to calculate the prevalence of ITs in the Italian pediatric population and to assess frequency and genotype–phenotype correlation of different types of mutations in our study cohort. Patients with inherited thrombocytopenia aged from 0 to 18 years in which the genetic defects have been identified and/or with a suggestive familiar history of thrombocytopenia were included in the study. Patients with thrombocytopenia due to other causes (e.g., neonatal, immune, oncological, or infective), aged over 18 years, and without an identifiable genetic defect and a suggestive familiar history of thrombocytopenia were excluded. Demographic data, family history, genetic variant, clinical characteristics, treatments, and laboratory findings were collected. Bleeding scores were assigned by clinicians using the Buchanan and Adix scoring system, as this is routinely used by our Italian working group (14). Thrombocytopenia was defined as a platelet count <150 × 109/L. A platelet count between 150 and 450 × 109/L was considered normal (15, 16). Genetic variants were classified by testing laboratories as pathogenic, likely pathogenic, variants of uncertain significance (VUS), likely benign, or benign/polymorphism, following the 2007 guidelines from the American College of Medical Genetic and Genomics (17).
Local ethics committee approval and written informed consent were obtained. The caregivers provided signed consent forms and data collection was performed according to Italian regulation for personal data protection.
The patients described in this manuscript were screened by different centers over a time span of nearly 20 years. In addition, the technologies used to screen for mutations have changed profoundly over the course of this study. Most patients enrolled between 2003 and 2017 were analyzed by Sanger sequencing with candidate gene approach based on the clinical characteristics of the patient and after application of the diagnostic algorithm described by Balduini et al. (18).
Since 2017, most patients were analyzed by the Medical Genetics Unit of IRCCS Burlo Garofolo Children's Hospital in Trieste by target sequence approach based on Ion Torrent Personal Genome Machine (Ion PGMTM) platform. Sequencing primers were designed on the coding and intronic flanking regions of 28 IT genes using the Ion Ampliseq Designer software (https://www.ampliseq.com) (Supplementary Table S2).
Following the manufacturer's recommendations (Life Technologies), two multiplex PCRs were carried out for each sample using the Ion AmpliSeq library kit 2.0. Emulsion-PCR and enrichment reactions were performed on the template using Ion One Touch 2 system. Sequencing reactions were performed using Ion PGMTM Sequencing 200 Kit v2. Sequencing data were analyzed using Ion Torrent Suite software (v.5.12). Data were aligned with hg19 human genomic sequence using the plug-in Variant Caller (TSVC v5.6 and v.5.12). Functional annotations of all the sequence variants were performed using the wANNOVAR software (http://wannovar.usc.edu/).
Only rare variants with a minor allele frequency <0.01 were considered for the analysis, and all variants reported were confirmed by Sanger sequencing using standard conditions in an ABI 3100 automated sequencer (Applied Biosystems, Foster City, CA, United States). Some patients underwent whole exome sequencing (WES) analyses.
The large amount of genetic data emerging from next-generation sequencing approaches has the consequence that the meaning attributed to identified variants can also change rapidly over time due to the frequency with which these variants are detected or by the fact that some variants are identified only in certain groups of individuals. Therefore, especially in cases where variants of uncertain significance have been identified, their interpretation must periodically be re-evaluated. This process is very important because it allows some variants to be reclassified over time.
All tools used to assess the pathogenicity of the variants identified in this work were again browsed and updated during the revision process of this manuscript in order to provide the most up-to-date interpretation. Variant pathogenicity was evaluated taking into account several criteria that do not always agree in classification. Specifically, it was assessed whether the variant had been functionally studied or had already been reported in the literature and/or classified as a cause of disease by the Human Gene Mutation Database (HGMD). In addition, interpretations from the InterVar and ClinVar software were also considered.
A total of 139 children with IT (82 males and 57 females) were enrolled. ITs prevalence in Italy ranged from 0.7 per 100,000 children during 2010 to 2 per 100,000 children during 2022. The median time between the onset of thrombocytopenia and the diagnosis of ITs was 1 year (range 0–18 years). Family history of thrombocytopenia has been reported in 90 patients (65%). The median time between the onset of thrombocytopenia and the diagnosis of ITs was 1 year in patients with a family history of thrombocytopenia (range 0–18 years) and 4 years in patients without a family history of thrombocytopenia (range 0–18 years). There are no statistically significant differences between the groups (p value: 0.6). Table 1 lists the demographic and baseline features of the included patients. Among 139 children with ITs, in 73 (53%) children, almost one defective gene has been identified. In 61 patients, a pathogenic mutation has been identified (Table 2). Among them, two patients also carry a VUS, and four others harbor two VUS variants. VUS variants were identified in further eight patients (6%), four of which carry more than one variant VUS. Three patients (2%) had a likely pathogenic variant, while in one patient (1%), a variant was identified that was initially given an uncertain significance but was later classified as benign (32). In addition, in 17 patients, the genetic diagnosis is not available, but their family history and clinical/laboratory features strongly suggest the presence of a specific genetic cause. In 49 children (35%), no genetic defects were identified. These results are summarized in Figure 1.
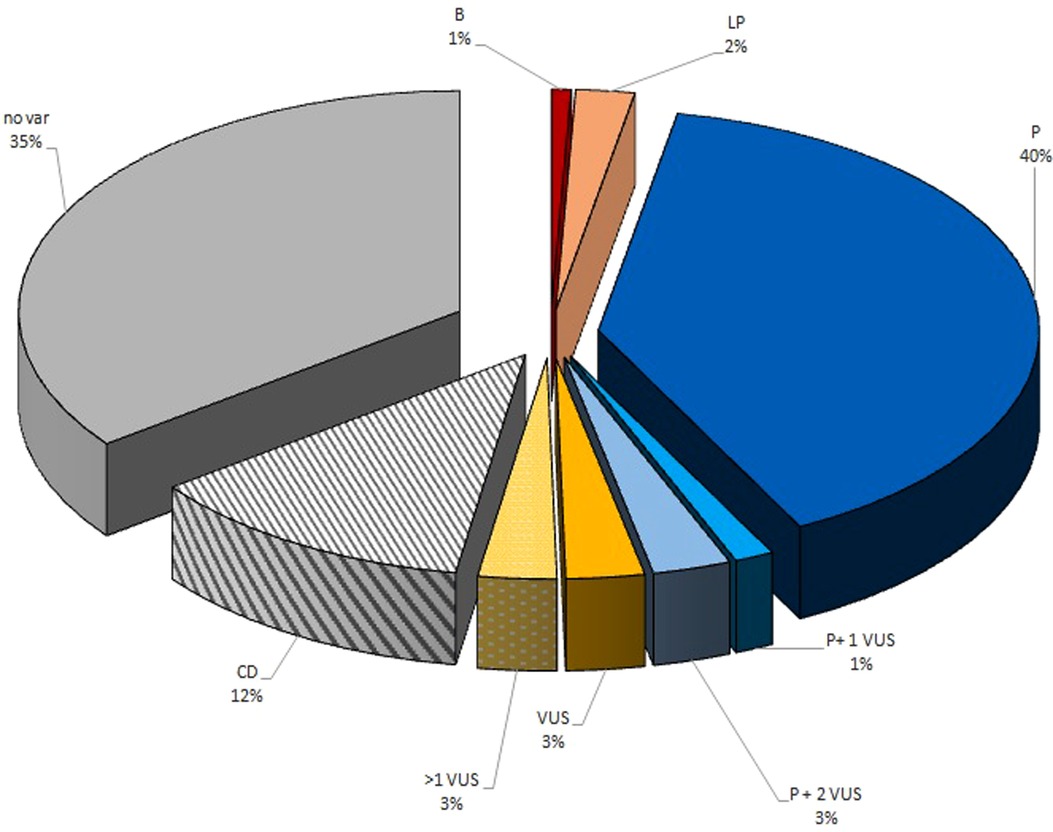
Figure 1. Graphical representation of IT genetic variants detected on the CHIPS cohort. B, benign variant; LP, likely pathogenetic variant; PV, pathogenetic variant; VUS, variant of uncertain significance; CD, clinical diagnosis based on clinical features or family history; no var, no candidate variants identified.

Table 1. Demographic and baseline features of the included patients.
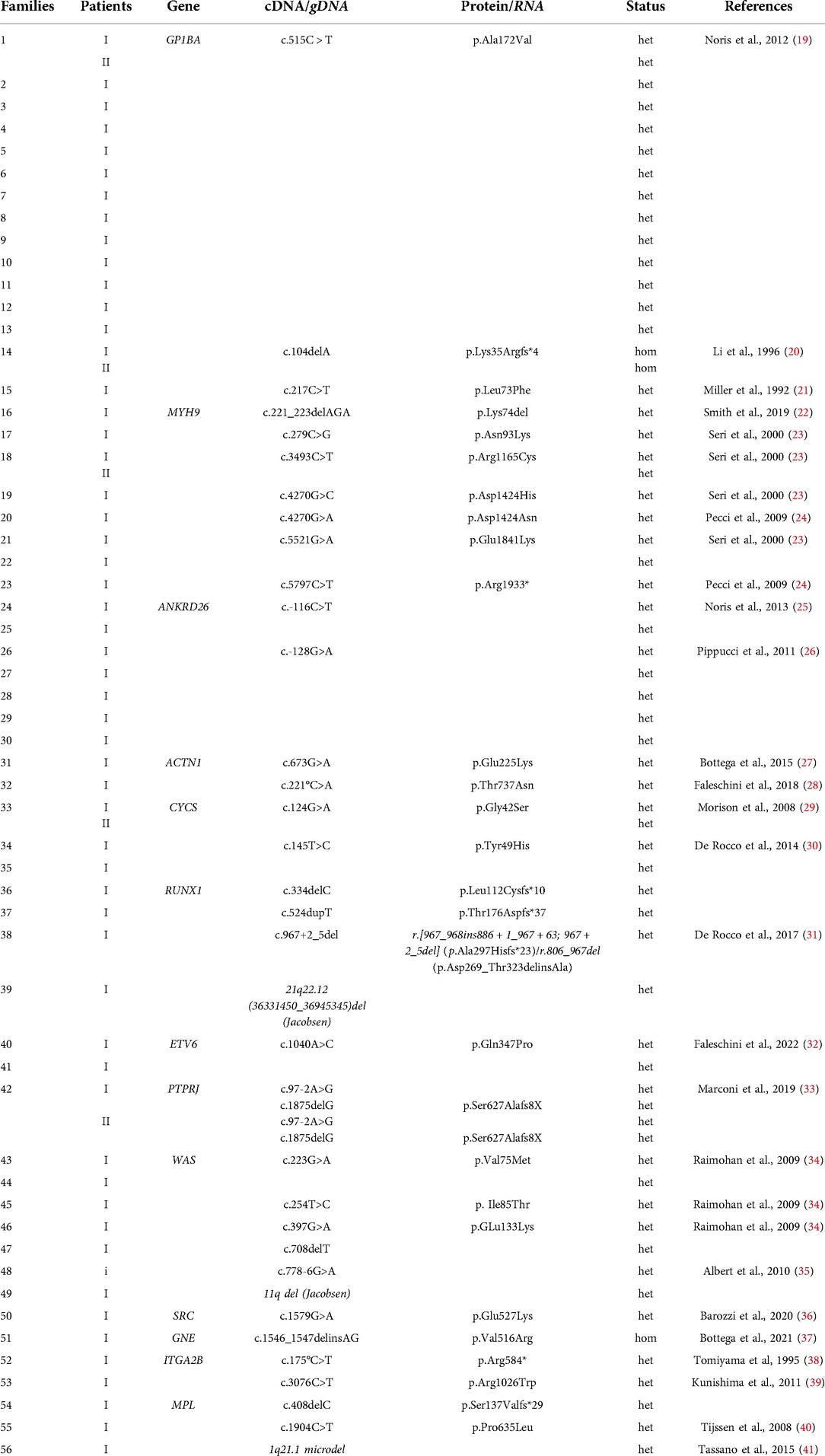
Table 2. Patients with pathogenetic variants.
Children’s median bleeding score was 0 (range 0–4). In ninetyseven patients (70%), thrombocytopenia was not associated with other clinically apparent disorders. However, 42 children (30%) had one or more additional clinical alterations. Immune disorders and/or recurrent infections (9%), cognitive impairment (8%), skeletal (4%) or otolaryngological abnormalities (5%), central nervous (4%) or cardiovascular system (4%) malformations, gastrointestinal (9%), dermatological (4%), ocular (3%), urogenital (5%), and endocrinological disorders (8%) were often associated with thrombocytopenia. Patients with pathogenic variants are described below.
Known mutation in the GP1BA gene was identified in 17 patients (12%) (7 males and 10 females). Among them, 14 patients from 13 families carry the c.515C>T (p.Ala172Val) mutation, also known as Bolzano mutation (19). In one of these patients (2-I), the Bolzano mutation was associated with the ABCG8 variant classified as VUS. With regard to the remaining three patients, a homozygous small deletion (c.104delA), which has been previously associated with the Bernard–Soulier syndrome (BSS) (20), was identified in two individuals belonging to same family. In accordance with a diagnosis of BSS, these patients show increased mean platelet volume (MPV) as well as increased bleeding tendency (Supplementary Table S1). The pathogenic variant c.217C>T (p.Leu73Phe) was identified in the last patient as described in Table 2. The median age at the diagnosis was 3 years (0–13 years), while the median platelet count at the diagnosis was 93 × 109/L (14–147 × 109/L). The median bleeding score was 0 (range 0–3). Three patients (18%) (14-I, 14-II, 13-I) required one or more treatments (e.g., red cell concentrates transfusions, platelet transfusions, tranexamic acid). No patient (94%) had symptoms or laboratory abnormalities associated with thrombocytopenia except one patient (6%) affected by craniofacial dysmorphisms (Goldenhar syndrome) (2-I).
Thirteen patients (9%), 10 males and 3 females, were affected by MYH9-related thrombocytopenia. Pathogenic mutations in the MYH9 gene were identified only in nine patients from eight different families (Table 2). In one patient (17-I), mutation of MYH9 was associated with two VWF variants classified as VUS. Although in the remaining four patients the genetic diagnosis is not available, their family history and clinical/laboratory features allowed us to include them in the MYH9-related thrombocytopenia patients. The median age at the diagnosis was 5 years (0–13 years) while the median platelet count at the diagnosis was 50 × 109/L (9–90 × 109/L). The median bleeding score was 1 (range 0–3). Three patients (23%) required one or more treatments. Among 13 patients with MYH9-related thrombocytopenia, 10 patients (77%) were asymptomatic, 1 patient (8%) suffered from sensorineural hearing loss, 1 patient (8%) had chronic renal failure and neurobehavioral disorders (19-I), and 1 patient (18-I) (8%) had facial dysmorphisms and Hirschsprung’s disease.
ANKRD26-related thrombocytopenia was detected in nine patients (6%) (five males and four females). Although a known mutation was identified in seven patients (Table 2), the family history (presence of the genetic mutation in the family, thrombocytopenia inherited with an autosomal dominant pattern, and history of myelodysplastic syndrome and/or myeloid neoplasms) and clinical/laboratory features of the other two patients strongly suggest the presence of a genetic cause. In one patient (30-I) without significant clinical and laboratory characteristic, ANKRD26 mutation was associated with GP1BA and NBEAL2 variants classified as VUS. Among patients with ANKRD26-related thrombocytopenia, the median age at the diagnosis was 7 years (3–15 years) while the median platelet count at the diagnosis was 58 × 109/L (41–115 × 109/L). The median bleeding score was 0 (range 0–3). Four patients (44%) (29-I, 25-I, 24-I) required one or more treatments. Among nine patients with ANKRD26-related thrombocytopenia, one patient (25-I) (11%) developed myelodysplasia at the age of 10. Exitus occurred after hematopoietic stem cell transplantation (HSCT) owing to complications of severe idiopathic pneumonia syndrome.
WAS-related thrombocytopenia was detected in 12 patients (7%) (eleven males and one female). Among them only six patients obtained a molecular diagnosis (Table 2), while in six patients for whom the genetic data are not available, the diagnosis was based on their family history and clinical/laboratory features. The median age at the diagnosis was 4 years (0–8 years) while the median platelet count at the diagnosis was 29 × 109/L (13–63 × 109/L). The median bleeding score was 0 (range 0–3). Ten patients (83%) required one or more treatments. Among 12 patients with WAS-related thrombocytopenia, 8 patients (66%) had no symptoms or laboratory abnormalities associated with thrombocytopenia. One patient (48-I) (8%) was affected by splenomegaly, immunodeficiency, otolaryngological alterations, and central nervous abnormalities; another patient (8%) developed dermatological alterations, ischemic stroke at the age of 4 years, and hepatic lymphoblastic lymphoma at the age of 9 years. He had undergone HSCT with complete remission of the disease. In addition, one patient (47-I) (9%) at birth had experienced cephalohematoma and cholestasis. He had undergone HSCT at the age of 6 months and died of complications. Two patients (44-I) (16%) had undergone HSCT at the age of 2 years with complete remission of the disease.
ACTN1-related thrombocytopenia was diagnosed in three male patients (2%). As reported in Table 2, two of them carry a known pathogenic mutation, while in the remaining patient, the diagnosis was possible due to the clinical/laboratory features and because the diagnosis of ACTN1-RT was reported in the patient's father. The median age at the diagnosis was 3 years (2–11 years) while the median platelet count at the diagnosis was 97 × 109/L (87–104 × 109/L). In all patients, bleeding symptoms were mild (median bleeding score 1, range 0–1). Among three patients with ACTN1-related thrombocytopenia, one patient (33%) was asymptomatic. One patient (33%) had a congenital abnormality of the urinary tract (vesicoureteral reflux), while one patient (32-I) (33%) had splenomegaly and gallbladder anomalies.
A known pathogenic mutation in the CYCS gene was identified in four male patients (3%), two of them belonging to the same family (Table 2). The median age at the diagnosis was 10 years (3–18 years) while the median platelet count at the diagnosis was 124 × 109/L (103–144 × 109/L). All children presented mild bleeding symptoms without additional symptoms or laboratory abnormalities associated with thrombocytopenia.
RUNX1 mutations were identified in four patients (3%) (two males and two females). Of note, mutation c.967+2_5del, generates three different RNA transcripts and two different protein products as reported in Table 2. The pathogenetic effect of this mutation is reported in detail by De Rocco and colleagues as well as the association of c.967+2_5del with impaired platelet aggregation (30). Defects in platelet aggregation were also observed in patient 39-I in whom a deletion of about 600 kb, resulting in the lack of exons 1 and 2 of the RUNX1 gene, was detected. The median age at the diagnosis was 6 years (3–9 years) while the median platelet count at the diagnosis was 84 × 109/L (44–141 × 109/L). None of the patients reported bleeding events (median bleeding score: 0, range 0–0). Among four patients with RUNX1-related thrombocytopenia, two patients (50%) had not abnormalities associated. Thrombocytopenia, cognitive impairment, and thyroid dysfunction were reported in one patient (37-I) (25%). In addition, at the age of 17, one patient (39-I) (25%) developed a myelodysplastic syndrome, a successively acute myeloid leukemia. She had undergone HSCT with complete remission of the disease.
ETV6-related thrombocytopenia was detected in two patients (1%) (one male and one female). Of note, the two patients, belonging to two unrelated families, carry the same mutation, recently classified as pathogenic (32). The median age at the diagnosis was 1 year (0–12 years) while the median platelet count at the diagnosis was 61 × 109/L (17–97 × 109/L). The median bleeding score was 0 (range 0–0). Both patients required one or more treatments. One patient (41-I) (50%) had cognitive impairment, epilepsy, and dyslexia.
Whole exome sequencing performed in two siblings (1%) (9-year-old male and 17-year-old female) allowed us to identify two compound heterozygous mutations in PTPRJ gene, responsible for a novel form of thrombocytopenia (33). The median platelet count at the diagnosis was 86 × 109/L (77–96 × 109/L). The girl had a history of spontaneous bleedings consisting in menorrhagia, easy bruising, petechiae, and epistaxis, resulting in mild iron deficiency anemia. The boy also presented spontaneous bleeding though of a milder degree. Except for bleeding tendency, medical history of both probands was unremarkable, and physical examination did not reveal any relevant abnormalities.
Genetic analysis allowed us to identify six further pathogenetic mutations in other five thrombocytopenia related genes, which are shown in Table 2 and are described as follows.
11q23 deletion (Jacobsen syndrome) was found in a 3-year-old girl with multiple cardiovascular and skeletal abnormalities and vascular malformations (aberrant subclavian artery). In this patient, thrombocytopenia was mild (100 × 109/L at the diagnosis) without bleeding symptoms (Bleeding score: 0).
The pathogenetic microdeletion 1q21.1, associated with the thrombocytopenia-absent radius (TAR) syndrome, was detected in heterozygous state in a new-born with multiple skeletal, otolaryngological, and ocular anomalies and severe bleeding tendency. The TAR syndrome is a recessive form of thrombocytopenia caused by the combination of the 1q21.1 microdeletion in association with specific pathogenic single nucleotide polymorphisms (SNPs) in the other allele of the RBM8A gene; therefore, the alteration identified is not sufficient to obtain a molecular diagnosis. However, the clinical phenotype of this patient strongly suggests the presence of the second pathogenic mutation in the RBM8A gene that has not yet been identified.
Thrombocytopenia caused by a mutation in the SRC tyrosine kinase gene was found in an infant who suffered intracranial hemorrhage during birth. At 3 years of age, he developed myelodysplasia and had undergone HSCT complicated by acute graft versus host disease.
In patient 51-I reported in Table 2, the homozygous c.1546_1547delinsAG mutation in the GNE gene was associated with severe thrombocytopenia (median platelet count: 5 × 109/L) and moderate hemorrhagic phenotype (bleeding score: 2).
The pathogenetic c.3076C>T (p.Arg1026Trp) mutation in the ITGA2B gene was detected in one patient with a mild thrombocytopenia (median platelet count: 90 × 109/L) and hemorrhagic phenotype (bleeding score: 1). Of note, this patient also carried two VWF variants classified as VUS.
The pathogenetic c.175°C>T (p.Arg584*) mutation in the ITGA2B gene was detected in one patient (52-I) with a moderate thrombocytopenia (median platelet count: 18 × 109/L) and with significant bleeding episodes (bleeding score: 2).
Finally, MPL mutations (55-I) were identified in one patient who developed trilinear cytopenia and undergone HSCT at the age of 1 year with complete remission of the disease and in another one with thyroiditis and mild thrombocytopenia (median platelet count: 63 × 109/L). In the latter patient, MPL mutation (54-I) was detected together with a NBEAL2 variant classified as VUS.
In all patients, at least one clinical and laboratorial control was performed annually (range, 1–3). Follow-up valuation included (1) clinical assessment (100%); (2) complete blood count and reticulocyte count (100%); (3) peripheral blood smear (25%); (4) biochemical measurements of renal and hepatic function, electrolytes, and serum protein (20%); (5) urinalysis (5%); and (6) immunological assessment (3%), audiological evaluation (2%), and bone marrow examination (4%). The median time of follow-up was 2 years and 4 months (range, 0–16). Overall, 50 patients (35%) developed mucosal bleeding (22%) and cutaneous bleeding (13%), 4 patients (3%) developed myelodysplasia, and 7 patients (5%) had undergone HSCT. Among these patients, four patients had a complete remission of the disease, one patient had an acute graft vs. host disease, and two children died of complications.
ITs have been considered for a long time extremely rare diseases characterized by severe and life-threatening hemorrhagic symptoms. In the last few decades, considerable progress has been made in the understanding pathophysiology and molecular basis of ITs. To date, a total of 45 different forms of ITs have been identified with different clinical expression and variable prognosis (15, 42, 43). However, ITs are poorly known by clinicians and often misdiagnosed with most common forms of thrombocytopenia. The complexity of laboratory investigations available in few centers and the limited clinical experience in the knowledge of these forms are the most common causes of delayed diagnosis. Making an incorrect diagnosis exposes many patients to a suboptimal clinical management and useless therapies (44). Moreover, several forms of ITs predispose to additional complications, such as hematological malignancies or renal failure, which can be avoided with appropriate and timely treatment (45–49). Our retrospective–prospective multicenter study was the first study conducted in a large cohort of children of Italian population with the aim of improving knowledge about ITs. We found that the prevalence of ITs in the Italian pediatric population is higher with respect to previous data (4) and has increased significantly during the last few years. The progressive increase in the annual prevalence of ITs could be related to the improvement in knowledge of these disorders and the better ability to precociously identify them. In accordance with the possibility that diagnosis may be frequently complex and delayed, we found that the median time between the initial finding of thrombocytopenia and the diagnosis of inherited form was widely variable. The presence of a family history of thrombocytopenia, atypical features on the blood film, or associated diseases could lead to a prompt diagnosis (49). Although the median time to diagnosis appears lower in the group of patients with a family history of ITs compared to the group of patients without a family history, this difference is not statistically significant. This could be explained by the fact that patients with a familiar history of thrombocytopenia and a slightly lower-than-normal platelet count could consider medical intervention unnecessary and superfluous. Platelet count, genetic diagnosis, and clinical presentation varied considerably among the patients studied, which is consistent with the variability observed in the spectrum of ITs (42). We found that GP1BA-, MYH9-, ACTN1-, and ANKRD26-related thrombocytopenias are the most frequent diseases diagnosed with a highly variable clinical course and long-term prognosis. Their clinical and laboratory features are well recognized. According to several studies (50–52), we detected that in most patients, the mutation of GP1BA is exclusively associated with a macrothrombocytopenia. However, we reported in one patient multiple cardiovascular and skeletal dysmorphisms. Recently, Souto Filho et al. reported a case of Bolzano mutation associated with clinical features of 22q11.2 deletion syndrome with phenotypic spectrum of DiGeorge syndrome. This association could be explained by the fact that the constitutional hemizygosity of 22q11.2 may unmask an autosomal recessive disorder caused by alterations of the nondeleted GP1BA allele (53). Furthermore, we described in patients with MYH9-related thrombocytopenia neurobehavioral disorders, Hirschsprung’s disease, and facial dysmorphisms. In addition, congenital abnormality of the urinary tract, cardiac valve diseases, splenomegaly, and gallbladder anomalies were reported in two patients affected by ACTN1-related thrombocytopenia. The clinical significance of these abnormalities and their correlation with underlying platelet defect is still unknown, and future follow-up will be required.
As several forms of ITs are characterized by enlarged platelets, it is commonly recognized that the evaluation of platelet size is an important tool to suspect these diseases. In our study, we found that the MPV reported by the different centers is widely variable and not entirely correlated with the expected platelet size. However, the measurements of platelet size in ITs present substantial difficulties. As reported by Noris et al., the sensitivity and specificity of MPV in establishing platelet size is greatly variable depending on the instrument used. Furthermore, some of ITs (i.e., MYH9 and Bernard–Soulier syndrome) may present platelets that, due to their increased size, are unrecognized by the electronic counter, which therefore underestimates the MPV (54). In the last few years, new genes and de novo mutations responsible for inherited thrombocytopenia are continuously detected, and the classification of hereditary thrombocytopenias is updated constantly (55, 56). Therefore, pathogenicity could be due to different predisposing genetic variants in a polygenic setting. The use of next-generation sequencing (NGS) like whole genome and WES allowed the identification of causal genetic variants in both well-known and new genes involved in ITs, for example, SLFN14, FYB, STIM1, GFI1B, ETV6, and PTPRJ, but molecular mechanisms of some variants still remain unclear (31, 33, 57, 58). Although our knowledge regarding the causes of IT continues to grow, a genetic diagnosis is only reported in approximately 50% of patients and frequently variants of uncertain significance are detected (59). Recently, Johnson et al. analyzed 31 patients with ITs using whole exome sequencing. A variant of uncertain significance was identified in 51% of patients, while in 23% of patients, no variants have been detected (60). According to these studies, we observed that in less than half of IT patients, a pathogenic genetic defect have been identified. In addition, we reported that in 13% of patients, variants of uncertain significance have been detected, while in 33% of patients, no genetic defects were identified. Among variants of uncertain significance, we detected that NBEAL2 gene mutation is the most frequent. We found highly variable clinical pictures in these patients with a wide variety of diseases associated with thrombocytopenia such as splenomegaly, central nervous system involvement, behavior impairment, and progressive bone marrow alterations. The molecular mechanism that explains the variable clinical presentation still remains undefined. Bottega et al. compared the clinical features of 11 patients with gray platelets syndrome. In these patients, a worse clinical course was seen in individuals with biallelic NBEAL2 mutation. Moreover, in more than half of the patients, no gene alterations were identified suggesting that other defects in alternative genetic pathway are responsible for their platelet phenotype (61, 62). According to these studies, we found that in numerous patients, a genetic mutation was not detected or partially explained the pathogenic mechanism, although the clinical picture and anamnestic features are indicative of ITs.
Although further investigations are required to identify the genetic variations responsible for thrombocytopenia, the benefit of distinguishing ITs from acquired forms could become a critical step in improving patient clinical management and follow-up (63–66). Close monitoring including periodic clinical and laboratory examination could provide clinicians with greater knowledge of forms of thrombocytopenia without genetic identification.
In conclusion, our study provides a descriptive collection of the diagnosis of ITs in the pediatric Italian population. Despite the rarity of these hereditary disorders, collecting clinical and laboratory data and following patients over time could increase the knowledge of ITs and allow clinicians to diagnose them promptly and avoid further complications.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by Comitato Etico Locale, Policlinico di Bari, Bari, Italy. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
GL conceived the work and coordinated the study. MF and AS conducted the genetic investigations. VP wrote a draft. All authors approved the draft and finalized the paper. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.967417/full#supplementary-material.
ITs, inherited thrombocytopenias; CHIPS, “CHildren with Inherited Platelet disorders Surveillance” study; AIEOP, Italian Association of Pediatric Hematology and Oncology; VUS, variants of uncertain significance; HSCT, hematopoietic stem cell transplantation; BSS, Bernard–Soulier syndrome; MPV, mean platelet volume; NGS, next-generation sequencing; WES, whole exome sequencing
1. Israels SJ, El-Ekiaby M, Quiroga T, Mezzano D. Inherited disorders of platelet function and challenges to diagnosis of mucocutaneous bleeding. Haemophilia. (2010) 16(Suppl 5):152–9. doi: 10.1111/j.1365-2516.2010.02314.x
2. Bourguignon A, Tasneem S, Hayward CP. Screening and diagnosis of inherited platelet disorders. Crit Rev Clin Lab Sci. (2022) 59(6):405–44. doi: 10.1080/10408363.2022.2049199
3. Warren JT, Di Paola J. Genetics of inherited thrombocytopenias. Blood. (2022) 139(22):3264–77. doi: 10.1182/blood.2020009300
4. Israels SJ, Kahr WH, Blanchette VS, Luban NL, Rivard GE, Rand ML. Platelet disorders in children: a diagnostic approach. Pediatr Blood Cancer. (2011) 56(6):975–83. doi: 10.1002/pbc.22988
5. Palma-Barqueros V, Revilla N, Sánchez A, Zamora Cánovas A, Rodriguez-Alén A, Marín-Quílez A, et al. Inherited platelet disorders: an updated overview. Int J Mol Sci. (2021) 22(9):4521. doi: 10.3390/ijms22094521
6. Kozubik KS, Radova L, Reblova K, Smida M, Zaliova Kubricanova M, Baloun J, et al. Functional analysis of germline ETV6 W380R mutation causing inherited thrombocytopenia and secondary acute lymphoblastic leukemia or essential thrombocythemia. Platelets. (2021) 32(6):838–41. doi: 10.1080/09537104.2020.1802416
7. Boeckelmann D, Glonnegger H, Sandrock-Lang K, Zieger B. Pathogenic aspects of inherited platelet disorders. Hamostaseologie. (2021) 41(6):460–8. doi: 10.1055/a-1665-6249
8. Almazni I, Stapley R, Morgan NV. Inherited thrombocytopenia: update on genes and genetic variants which may be associated with bleeding. Front Cardiovasc Med. (2019) 6:80. doi: 10.3389/fcvm.2019.00080
9. Li X, Li Y, Lei M, Tian J, Yang Z, Kuang S, et al. Congenital thrombocytopenia associated with GNE mutations in twin sisters: a case report and literature review. BMC Med Genet. (2020) 21(1):224. doi: 10.1186/s12881-020-01163-2
10. Minkov M, Zeitlhofer P, Zoubek A, Kager L, Panzer S, Haas OA. Novel compound heterozygous mutations in two families with Bernard-Soulier syndrome. Front Pediatr. (2021) 8:589812. doi: 10.3389/fped.2020.589812
11. Abdelmoumen K, Fabre M, Ducastelle-Lepretre S, Favier R, Ballerini P, Bordet JC, et al. Eltrombopag for the treatment of severe inherited thrombocytopenia. Acta Haematol. (2021) 144(3):308–13. doi: 10.1159/000509922
12. Lassandro G, Palladino V, Vecchio GCD, Palmieri VV, Corallo PC, Faienza MF, et al. Thrombopoietin receptor agonists in children with immune thrombocytopenia: a new therapeutic era. Endocr Metab Immune Disord Drug Targets. (2021) 21(3):397–406. doi: 10.2174/1871530320666200531142244
13. Giordano P, Lassandro G, Barone A, Cesaro S, Fotzi I, Giona F, et al. Use of eltrombopag in children with chronic immune thrombocytopenia (ITP): a real life retrospective multicenter experience of the Italian Association of Pediatric Hematology and Oncology (AIEOP). Front Med. (2020) 7:66. doi: 10.3389/fmed.2020.00066
14. Buchanan GR, Adix L. Grading of hemorrhage in children with idiopathic thrombocytopenic purpura. J Pediatr. (2002) 141(5):683–8. doi: 10.1067/mpd.2002.128547
15. Nurden AT, Nurden P. Inherited thrombocytopenias: history, advances and perspectives. Haematologica. (2020) 105(8):2004–19. doi: 10.3324/haematol.2019.233197
16. Rokkam VR, Kotagiri R. Secondary thrombocytosis. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing (2022).
17. Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, et al. Molecular subcommittee of the ACMG laboratory quality assurance committee. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet Med. (2008) 10:294–300. doi: 10.1097/GIM.0b013e31816b5cae
18. Balduini CL, Cattaneo M, Fabris F, Gresele P, Iolascon A, Pulcinelli FM, et al. Inherited thrombocytopenias: a proposed diagnostic algorithm from the Italian Gruppo di Studio delle Piastrine. Hematologica. (2003) 88:582–92.
19. Noris P, Perrotta S, Bottega R, Pecci A, Melazzini F, Civaschi E, et al. Clinical and laboratory features of 103 patients from 42 Italian families with inherited thrombocytopenia derived from the monoallelic Ala156Val mutation of GPIbα (Bolzano mutation). Haematologica. (2012) 97(1):82–8. doi: 10.3324/haematol.2011.050682
20. Li C, Pasquale DN, Roth GJ. Bernard-Soulier syndrome with severe bleeding: absent platelet glycoprotein Ib alpha due to a homozygous one-base deletion. Thromb Haemost. (1996) 76(5):670–4. doi: 10.1055/s-0038-1650640
21. Miller JL, Lyle VA, Cunningham D. Mutation of leucine-57 to phenylalanine in a platelet glycoprotein Ib alpha leucine tandem repeat occurring in patients with an autosomal dominant variant of Bernard-Soulier disease. Blood. (1992) 79(2):439–46. doi: 10.1182/blood.V79.2.439.439
22. Smith AS, Pal K, Nowak RB, Demenko A, Zaninetti C, Da Costa L, et al. MYH9-related disease mutations cause abnormal red blood cell morphology through increased myosin-actin binding at the membrane. Am J Hematol. (2019) 94(6):667–77. doi: 10.1002/ajh.25472
23. Seri M, Cusano R, Gangarossa S, Caridi G, Bordo D, Lo Nigro C, et al. Mutations in MYH9 result in the May-Hegglin anomaly, and Fechtner and Sebastian syndromes. The May-Heggllin/Fechtner syndrome consortium. Nat Genet. (2000) 26(1):103–5. doi: 10.1038/79063
24. Pecci A, Malara A, Badalucco S, Bozzi V, Torti M, Balduini CL, et al. Megakaryocytes of patients with MYH9-related thrombocytopenia present an altered proplatelet formation. Thromb Haemost. (2009) 102(1):90–6. doi: 10.1160/TH09-01-0068
25. Noris P, Favier R, Alessi MC, Geddis AE, Kunishima S, Heller PG, et al. ANKRD26-related thrombocytopenia and myeloid malignancies. Blood. (2013) 122(11):1987–9. doi: 10.1182/blood-2013-04-499319
26. Pippucci T, Savoia A, Perrotta S, Pujol-Moix N, Noris P, Castegnaro G, et al. Mutations in the 5’ UTR of ANKRD26, the ankirin repeat domain 26 gene, cause an autosomal-dominant form of inherited thrombocytopenia, THC2. Am J Hum Genet. (2011) 88(1):115–20. doi: 10.1016/j.ajhg.2010.12.006
27. Bottega R, Marconi C, Faleschini M, Baj G, Cagioni C, Pecci A, et al. ACTN1-related thrombocytopenia: identification of novel families for phenotypic characterization. Blood. (2015) 125(5):869–72. doi: 10.1182/blood-2014-08-594531
28. Faleschini M, Melazzini F, Marconi C, Giangregorio T, Pippucci T, Cigalini E, et al. ACTN1 mutations lead to a benign form of platelet macrocytosis not always associated with thrombocytopenia. Br J Haematol. (2018) 183(2):276–88. doi: 10.1111/bjh.15531
29. Morison IM, Cramer Bordé EM, Cheesman EJ, Cheong PL, Holyoake AJ, Fichelson S, et al. A mutation of human cytochrome c enhances the intrinsic apoptotic pathway but causes only thrombocytopenia. Nat Genet. (2008) 40(4):387–9. doi: 10.1038/ng.103
30. De Rocco D, Cerqua C, Goffrini P, Russo G, Pastore A, Meloni F, et al. Mutations of cytochrome c identified in patients with thrombocytopenia THC4 affect both apoptosis and cellular bioenergetics. Biochim Biophys Acta. (2014) 1842(2):269–74. doi: 10.1016/j.bbadis.2013.12.002
31. De Rocco D, Melazzini F, Marconi C, Pecci A, Bottega R, Gnan C, et al. Mutations of RUNX1 in families with inherited thrombocytopenia. Am J Hematol. (2017) 92(6):E86–8. doi: 10.1002/ajh.24703
32. Faleschini M, Ammeti D, Papa N, Alfano C, Bottega R, Fontana G, et al. ETV6-related thrombocytopenia: dominant negative effect of mutations as common pathogenic mechanism. Haematologica. (2022) 107(9):2249–54. doi: 10.3324/haematol.2022.280729
33. Marconi C, Di Buduo CA, LeVine K, Barozzi S, Faleschini M, Bozzi V, et al. Loss-of-function mutations in PTPRJ cause a new form of inherited thrombocytopenia. Blood. (2019) 133(12):1346–57. doi: 10.1182/blood-2018-07-859496
34. Rajmohan R, Raodah A, Wong MH, Thanabalu T. Characterization of Wiskott-Aldrich syndrome (WAS) mutants using Saccharomyces cerevisiae. FEMS Yeast Res. (2009) 9(8):1226–35. doi: 10.1111/j.1567-1364.2009.00581.x
35. Albert MH, Bittner TC, Nonoyama S, Notarangelo LD, Burns S, Imai K, et al. X-linked thrombocytopenia (XLT) due to WAS mutations: clinical characteristics, long-term outcome, and treatment options. Blood. (2010) 115(16):3231–8. doi: 10.1182/blood-2009-09-239087
36. Barozzi S, Di Buduo CA, Marconi C, Bozzi V, Seri M, Romano F, et al. Pathogenetic and clinical study of a patient with thrombocytopenia due to the p.E527K gain-of-function variant of SRC. Haematologica. (2021) 106(3):918–22. doi: 10.3324/haematol.2020.268516
37. Bottega R, Marzollo A, Marinoni M, Athanasakis E, Persico I, Bianco AM, et al. GNE-related thrombocytopenia: evidence for a mutational hotspot in the ADP/substrate domain of the GNE bifunctional enzyme. Haematologica. (2022) 107(3):750–4. doi: 10.3324/haematol.2021.279689
38. Tomiyama Y, Kashiwagi H, Kosugi S, Shiraga M, Kanayama Y, Kurata Y, et al. Abnormal processing of the glycoprotein IIb transcript due to a nonsense mutation in exon 17 associated with Glanzmann's thrombasthenia. Thromb Haemost. (1995) 74(2):811. doi: 10.1182/blood-2010-12-323691
39. Kunishima S, Kashiwagi H, Otsu M, Takayama N, Eto K, Onodera M, et al. Heterozygous ITGA2B R995W mutation inducing constitutive activation of the αIIbβ3 receptor affects proplatelet formation and causes congenital macrothrombocytopenia. Blood. (2011) 117(20):5479–84. doi: 10.1182/blood-2010-12-323691
40. Tijssen MR, di Summa F, van den Oudenrijn S, Zwaginga JJ, van der Schoot CE, Voermans C, et al. Functional analysis of single amino-acid mutations in the thrombopoietin-receptor Mpl underlying congenital amegakaryocytic thrombocytopenia. Br J Haematol. (2008) 141(6):808–13. doi: 10.1111/j.1365-2141.2008.07139.x
41. Tassano E, Gimelli S, Divizia MT, Lerone M, Vaccari C, Puliti A, et al. Thrombocytopenia-absent radius (TAR) syndrome due to compound inheritance for a 1q21.1 microdeletion and a low-frequency noncoding RBM8A SNP: a new familial case. Mol Cytogenet. (2015) 8:87. doi: 10.1186/s13039-015-0188-6
42. Pecci A, Balduini CL. Inherited thrombocytopenias: an updated guide for clinicians. Blood Rev. (2021) 48:100784. doi: 10.1016/j.blre.2020.100784
43. Pluthero FG, Kahr WHA. Recent advances in inherited platelet disorders. Curr Opin Hematol. (2019) 26(5):313–9. doi: 10.1097/MOH.0000000000000525
44. Balduini CL, Savoia A, Seri M. Inherited thrombocytopenias frequently diagnosed in adults. J Thromb Haemost. (2013) 11(6):1006–19. doi: 10.1111/jth.12196.
45. Balduini A, Raslova H, Di Buduo CA, Donada A, Ballmaier M, Germeshausen M, et al. Clinic, pathogenic mechanisms and drug testing of two inherited thrombocytopenias, ANKRD26-related thrombocytopenia and MYH9-related diseases. Eur J Med Genet. (2018) 61(11):715–22. doi: 10.1016/j.ejmg.2018.01.014
46. Iqbal NT, Li W. Pay attention to neutrophil inclusions in pediatric patients with thrombocytopenia. Blood. (2019) 134(11):907. doi: 10.1182/blood.2019002222
47. Savoia A, Pecci A. MYH9-related disease. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Mirzaa GM, Amemiya A, editors. Genereviews®. Seattle, WA: University of Washington, Seattle (2008). p. 1993–2022.
48. Ferrari S, Regazzo D, Omenetto E, Scaroni C, Semenzato G, Fabris F, et al. A novel RUNX1 mutation with ANKRD26 dysregulation is related to thrombocytopenia in a sporadic form of myelodysplastic syndrome. Aging Clin Exp Res. (2021) 33(7):1987–92. doi: 10.1007/s40520-020-01709-7
49. Giordano P, Lassandro G, Sangerardi M, Faienza MF, Valente F, Martire B. Autoimmune haematological disorders in two Italian children with Kabuki syndrome. Ita J Pediatr. (2014) 25(40):10. doi: 10.1186/1824-7288-40-10
50. Reisi N. Bernard-Soulier syndrome or idiopathic thrombocytopenic purpura: a case series. Caspian J Intern Med. (2020) 11(1):105–9. doi: 10.22088/cjim.11.1.105
51. Monteiro M, Almeida L, Morais M, Dias L. Bernard Soulier syndrome: a rare, frequently misdiagnosed and poorly managed bleeding disorder. BMJ Case Rep. (2021) 14(8):e243518. doi: 10.1136/bcr-2021-243518
52. Sandrock K, Knöfler R, Greinacher A, Fürll B, Gerisch S, Schuler U, et al. Novel mutation in Bernard-Soulier syndrome. Transfus Med Hemother. (2010) 37(5):278–84. doi: 10.1159/000320255
53. Souto Filho JTD, Ribeiro HAA, Fassbender IPB, Ribeiro JMMC, Ferreira Júnior WDS, Figueiredo LCS. Bernard-Soulier syndrome associated with 22q11.2 deletion and clinical features of DiGeorge/velocardiofacial syndrome. Blood Coagul Fibrinolysis. (2019) 30(8):423–5. doi: 10.1097/MBC.0000000000000849
54. Noris P, Klersy C, Gresele P, Giona F, Giordano P, Minuz P, et al.; Italian Gruppo di Studio delle Piastrine. Platelet size for distinguishing between inherited thrombocytopenias and immune thrombocytopenia: a multicentric, real life study. Br J Haematol. (2013) 162(1):112–9. doi: 10.1111/bjh.12349
55. Mekchay P, Ittiwut C, Ittiwut R, Akkawat B, Le Grand SM, Leela-Adisorn N, et al. Whole exome sequencing for diagnosis of hereditary thrombocytopenia. Medicine. (2020) 99(47):e23275. doi: 10.1097/MD.0000000000023275
56. Collins J, Astle WJ, Megy K, Mumford AD, Vuckovic D. Advances in understanding the pathogenesis of hereditary macrothrombocytopenia. Br J Haematol. (2021) 195(1):25–45. doi: 10.1111/bjh.17409
57. Brøns N, Zaninetti C, Ostrowski SR, Petersen J, Greinacher A, Rossing Met al.. Inherited thrombocytopenias frequently diagnosed in adults. J Thromb Haemost. (2013) 11(6):1006–19. doi: 10.1111/jth.12196
58. Cordero-Sanchez C, Pessolano E, Riva B, Vismara M, Trivigno SMG, Clemente N, et al. CIC-39Na reverses the thrombocytopenia that characterizes tubular aggregate myopathy. Blood Adv. (2022) 6(15):4471–84. doi: 10.1182/bloodadvances.2021006378
59. Balduini CL, Pecci A, Noris P. Inherited thrombocytopenias: the evolving spectrum. Hamostaseologie. (2012) 32(4):259–70. doi: 10.5482/ha12050001
60. Johnson B, Doak R, Allsup D, Astwood E, Evans G, Grimley C, et al. A comprehensive targeted next-generation sequencing panel for genetic diagnosis of patients with suspected inherited thrombocytopenia. Res Pract Thromb Haemost. (2018) 2(4):640–52. doi: 10.1002/rth2.12151
61. Bottega R, Pecci A, De Candia E, Pujol-Moix N, Heller PG, Noris P, et al. Correlation between platelet phenotype and NBEAL2 genotype in patients with congenital thrombocytopenia and α-granule deficiency. Haematologica. (2013) 98(6):868–74. doi: 10.3324/haematol.2012.075861
62. Pluthero FG, Di Paola J, Carcao MD, Kahr WHA. NBEAL2 mutations and bleeding in patients with gray platelet syndrome. Platelets. (2018) 29(6):632–5. doi: 10.1080/09537104.2018.1478405
63. Valente M, Cortesi PA, Lassandro G, Mathew P, Pocoski J, Molinari AC, et al. Health economic models in hemophilia A and utility assumptions from a clinician's perspective. Pediatr Blood Cancer. (2015) 62(10):1826–31. doi: 10.1002/pbc.25543
64. Giordano P, Lassandro G, Valente M, Molinari AC, Ieranò P, Coppola A. Current management of the hemophilic child: a demanding interlocutor. Quality of life and adequate cost-efficacy analysis. Pediatr Hematolo Oncol. (2014) 31(8):687–702. doi: 10.3109/08880018.2014.930768.
65. Faienza MF, Lassandro G, Chiarito M, Valente F, Ciaccia L, Giordano P. How physical activity across the lifespan can reduce the impact of bone ageing: a literature review. Int J Environ Res Public Health. (2020) 17(6):1862. doi: 10.3390/ijerph17061862
Keywords: inherited thrombocytopenia, platelet, bleeding diseases/disorders, children, congenital thrombocytopenia
Citation: Lassandro G, Palladino V, Faleschini M, Barone A, Boscarol G, Cesaro S, Chiocca E, Farruggia P, Giona F, Gorio C, Maggio A, Marinoni M, Marzollo A, Palumbo G, Russo G, Saracco P, Spinelli M, Verzegnassi F, Morga F, Savoia A and Giordano P (2022) “CHildren with Inherited Platelet disorders Surveillance” (CHIPS) retrospective and prospective observational cohort study by Italian Association of Pediatric Hematology and Oncology (AIEOP). Front. Pediatr. 10:967417. doi: 10.3389/fped.2022.967417
Received: 12 June 2022; Accepted: 18 October 2022;
Published: 22 November 2022.
Edited by:
Oktay Kirak, University of Freiburg, GermanyReviewed by:
Jose Rivera Pozo, Hospital Universitario Morales Meseguer, Spain© 2022 Lassandro, Palladino, Faleschini, Barone, Boscarol, Cesaro, Chiocca, Farruggia, Giona, Gorio, Maggio, Marinoni, Marzollo, Palumbo, Russo, Saracco, Spinelli, Verzegnassi, Morga, Savoia and Giordano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppe Lassandro Z2l1c2VwcGVsYXNzYW5kcm9AbGl2ZS5jb20=
Specialty Section: This article was submitted to Pediatric Hematology and Hematological Malignancies, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.