 Blanca Gordon
Blanca Gordon Víctor González-Fernández
Víctor González-Fernández Laura Dos-Subirà
Laura Dos-Subirà
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr. , 18 November 2022
Sec. Pediatric Cardiology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.965204
This article is part of the Research Topic Congenital Heart Disease: A Lifelong Chronic Condition View all 19 articles
Myocardial fibrosis resulting from the excessive deposition of collagen fibers through the myocardium is a common histopathologic finding in a wide range of cardiovascular diseases, including congenital anomalies. Interstitial fibrosis has been identified as a major cause of myocardial dysfunction since it distorts the normal architecture of the myocardium and impairs the biological function and properties of the interstitium. This review summarizes current knowledge on the mechanisms and detrimental consequences of myocardial fibrosis in heart failure and arrhythmias, discusses the usefulness of available imaging techniques and circulating biomarkers to assess this entity and reviews the current body of evidence regarding myocardial fibrosis in the different subsets of congenital heart diseases with implications in research and treatment.
The history of congenital heart disease (CHD) is a history of hope and success. Since the first ligation of a patent ductus arteriosus accomplished by Gross and Hubbard on August 8, 1938 (1), an unstoppable flow of innovative and creative surgical and interventional techniques has dealt with the most unimaginable cardiac malformations. Thousands of children destined to die soon after birth have been able to reach adulthood and, in most cases, develop a near-normal life. But the history of CHD is also paved with uncertainties. These repaired hearts are frequently afflicted with long-term complications, predominantly heart failure (HF) and arrhythmias. Our understanding of the mechanisms behind these complications and the available treatment options is based on the knowledge borrowed from acquired heart diseases and raises more questions than answers. The singularities of CHD should compel us to look beyond the ordinary and seek personalized, precision medicine for our patients.
During the last years, the myocardial interstitium (MI) has gone from the role of innocent bystander to a preeminent position in the pathogenesis of long-term cardiovascular complications, mainly HF. The MI is not a passive entity, but rather a complex and dynamic microenvironment that undergoes constant turnover: it ensures structural myocardial integrity, executes the repair response after injury, provides the means for force transmission throughout the cardiac cycle, translates myocyte shortening into overall ventricular pump function and facilitates communication between cells (2).
The accumulation of fibrillar collagen in the interstitial space, the so-called myocardial interstitial fibrosis, has been identified as a major cause of myocardial dysfunction. It not only distorts this normal architecture but also impairs its biological function and properties.
The term “interstitial myocardial disease” was first coined in 1989 by Weber (3) in reference to maladaptive effects on interstitial remodeling in response to altered hemodynamic loading conditions and/or myocardial damage. These changes, including fibroblast activation, formation of myofibroblasts, inflammation and altered expression of metalloproteinases, ultimately lead to myocardial fibrosis (MF) (4).
Altered hemodynamic conditions are a common feature of most CHDs, both before and after palliation or complete repair. Therefore, MF is expected to be one of the main maladaptative mechanisms behind the progressive deterioration of ventricular function in our patients and a potential therapeutic target.
This paper aims to summarize current knowledge about MF in terms of pathophysiology, diagnostic tools and potential treatments, focusing on the CHD population.
For a simplified description, the myocardium can be dichotomized into two components. The cellular component is mainly represented by cardiomyocytes, occupying 75% of the myocardium but only conforming one-third of the cell population. The rest is constituted of endothelial cells, vascular smooth muscle cells, fibroblasts, macrophages and mast cells.. The non-cellular component of the myocardium is also located in the interstitial space and mainly comprises fibrillar collagen (95% of the extracellular matrix) but has also other constituents: proteoglycans, glycosaminoglycans and different bioactive signaling molecules (2, 5).
Among the cellular constituents, fibroblasts are responsible for the synthesis and maintenance of cardiac connective tissue. Fibroblasts can migrate to areas of damaged myocardium (i.e., surgical injury or myocardial infarction) where they trans-differentiate into myofibroblasts, which are crucial cells for cardiac remodeling and fibrosis (6). A myriad of chemokines, cytokines and growth factors are secreted in the injured myocardium and stimulate the activation and differentiation of fibroblasts. These bioactive elements are produced in response to different stimuli, including mechanical stress (7). The healing process includes degradation of the damaged tissue and subsequent production, cross-linking and maturation of a new extracellular matrix (ECM). Although myofibroblasts are the major source of ECM, other cells, like monocytes, macrophages, cardiomyocytes and non-differentiated fibroblasts contribute to the process by secreting pro-fibrotic growth factors (8).
As for non-cellular components, fibrillar collagen is the cornerstone, with five isoforms in the myocardial interstitium: collagen type I, III, IV, V and VI. Type I collagen is the predominant, representing 80% of the total. It offers the greatest tensile strength (comparable to steel) and represents the major determinant of myocardial stiffness. Type III collagen has more elastic characteristics, representing 10% of collagens, and the other types(IV, V and VI) constitute the remaining 10% (9, 10).
From a histopathological point of view, two types of MF should be distinguished. The reparative, replacement or focal myocardial fibrosis (FMF) develops as a healing mechanism at the site of a previous myocardial injury (infarction or surgical scar). The necrotic tissue is replaced by collagen fibrils that are cross-linked to provide mechanical stability in response to cardiomyocyte loss. On the other hand, reactive diffuse myocardial fibrosis (DMF) is characterized by a diffuse deposition of excess fibrous tissue relative to the mass of cardiomyocytes. In this case, MF appears as an accumulation of collagen fibers between individual cardiomyocytes and cardiomyocyte fascicles and around the intramyocardial vessels. As a result, the highly organized architecture of the myocardial interstitium is replaced with a thickened, poorly organized structure (11, 12) that alters the mechanical properties of the myocardium, thus impairing cardiac function and electrical activity, and contributing to the development of HF and the poor outcomes that characterize this condition.
Both types of MF can concur in the same individual, as seen in patients with ischemic cardiomyopathy that exhibit FMF at the necrotic site and DMF in the distant myocardium of both ventricles (13). Indeed, DMF can be facilitated by systemic factors activated secondarily to the loss of cardiac function (such as neurohormonal activation) (14) or related to extracardiac comorbidities (such as inflammation associated with metabolic syndrome or chronic kidney disease (15).
Beyond the quantity of fibrous tissue, the composition and physicochemical properties of the fibers are important in DMF. As previously mentioned, type I collagen fibers exhibit the highest stiffness, therefore, the ratio between type I and type III collagen will have an impact on overall myocardial stiffness. On the other hand, the type of intermolecular covalent bonding among collagen fibrils mediated by enzymes such as lysyl oxidases (LOXs), the so-called collagen cross-linking, will influence the resistance to degradation by matrix metalloproteinases (MMPs) and will also have an impact on myocardial stiffness.
Finally, it is well known that, coming from different embryonic origins, the left ventricle (LV) and the right ventricle (RV) respond distinctly to normal and abnormal loading conditions (16). Therefore, differences in the pattern and type of MF developed by both ventricles are also expected. The collagen content of a healthy RV is higher than that of the LV (17) and the RV and LV have different matrix metalloproteinase and ECM protein-expression patterns (18). Under the same overloading conditions, the RV exhibits less ability to develop adaptative remodeling (19, 20) leading to dilatation and dysfunction. This is particularly relevant in CHD, where morphologically RVs are exposed to extreme overloading conditions such as sustaining the systemic circulation, sometimes as a single ventricle, severe pulmonary hypertension or chronic pulmonary regurgitation as in repaired tetralogy of Fallot.
Interstitial MF has classically been related to the development of diastolic impairment since an excessive accumulation of insoluble collagen in the ECM leads to an abnormally stiff myocardium (see Figure 1).
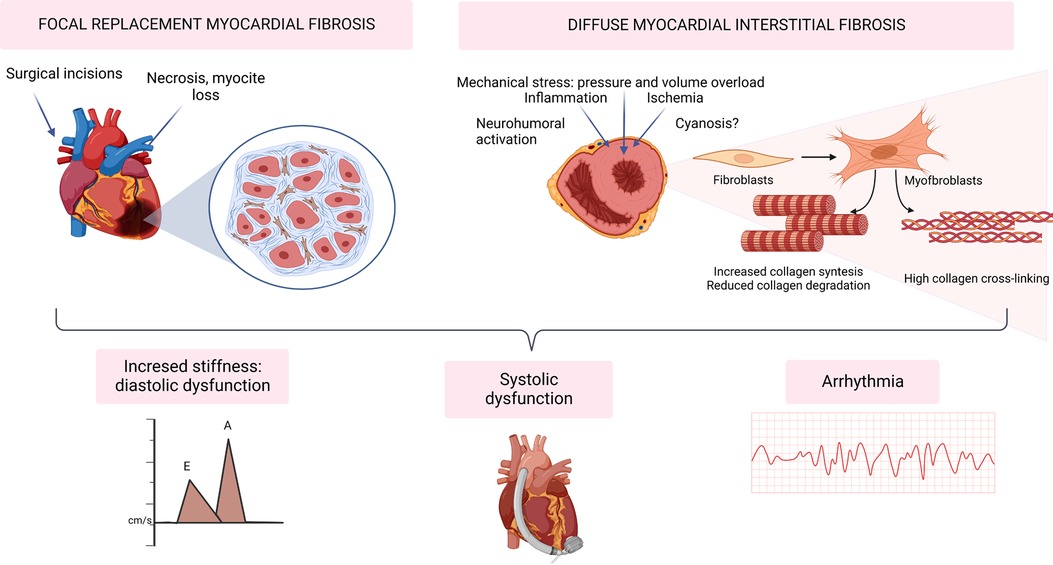
Figure 1. Myocardial fibrosis in the development of heart failure and arrhythmia in congenital heart disease. Created with BioRender.com.
In a physiological state, collagen fibers form a deformable elastic structure that stores energy when deformed from its neutral position during systolic contraction. Lengthening of the myocardium during diastole is then facilitated by the release of potential energy stored during systolic compression of these fibers, generating a suction effect that allows early diastolic filling (21).
When extracellular viscoelastic properties are impaired this suction effect is compromised. This leads to a higher dependence on atrial contraction and elevated filling pressures to maintain the stroke volume, which can also cause venous congestion and ultimately result in congestive heart failure.
Aside from the absolute amount of collagen, myocardial stiffness also depends on the characteristics of the fibers in the extracellular matrix. Thus, a higher fiber linkage and a higher proportion of type I collagen have also been correlated with impaired diastolic parameters in echocardiography, elevated peptides or elevated filling pressures in right heart catheterization (22–27).
In this setting, MF might just be part of the reparative response to the loss of contractile mass, becoming a marker of more advanced cardiomyopathy (28). However, the architecture of myocardial fibrillar collagen is also important in systolic performance, as it facilitates transduction of cardiomyocyte contraction (29, 30). Furthermore, the reduced stretching of the fibers in diastole can also compromise systolic function by altering the length-dependent cardiac muscle fiber shortening related to the Frank-Starling law.
Both FMF and DMF impair myocardial electrophysiology via several mechanisms, constituting an important arrhythmic substrate. Beyond re-entry tachycardias around macroscopic scars (e.g., surgical incisions), DMF interferes with myocardial electrophysiology by slowing down action potential propagation, initiating re-entry, promoting after-depolarizations, and increasing ectopic automaticity (31, 32).
Interestingly, some studies have shown that tachyarrhythmias can initiate or exacerbate MF by activation of profibrotic and proinflammatory signaling. This mechanism would perpetuate electric disturbances while contributing to the impairment of ventricular function (33–35).
Endomyocardial biopsy with histopathological analysis is the gold standard for MF diagnosis, but it presents several limitations as it is an invasive procedure and a difficult option in clinical practice (7, 36). Fortunately, currently available circulating and imaging biomarkers provide indirect MF diagnosis.
Cardiac magnetic resonance (CMR) methodologies for the detection of MF are based on the concept that fibrosis increases the extracellular space (37). As previously mentioned, MF can be divided into FMF, with dense macroscopic “replacement” fibrosis (scar) located to a specific, definable area; and DMF, which is more microscopic and uniform, with a global distribution and typically in response to chronic abnormal loading conditions (38–40).
There are two major CMR techniques used for MF assessment: late gadolinium enhancement (LGE) imaging and T1 mapping/extracellular volume (ECV) fraction calculation.
LGE is a CMR technique that identifies areas of discrete replacement fibrosis. Image acquisition is typically performed 10–20 min after administration of a gadolinium-based contrast agent. In damaged myocardial tissue, where there has been a significant localized expansion of the extracellular matrix, the distribution volume of the contrast agent is increased and the wash-out delayed (41).
LGE requires spatial heterogeneity to detect focal fibrosis, being a dichotomous method. It has the advantage that it analyzes the entire cardiac image, without focusing on a specific area, so it could be more sensitive for overall fibrosis burden, even though it requires a greater amount of fibrosis to be detectable visually (37).
ECV mapping quantifies MF by measuring the extracellular compartment depicted by the myocardial uptake of contrast relative to plasma (42). ECV represents the ratio of the interstitium relative to the total myocardial mass.
ECV has advantages over LGE, being intrinsically quantitative and able to assess both focal and diffuse fibrosis. However, although fibrosis is certainly a cause of increased ECV, it is not the only one, as extracellular space expansion may be due to other causes (myocardial vasculature, nonfibrillar proteins, edema, amyloidosis, etc.,) (7, 42, 43).
Even so, ECV is the most widely used imaging biomarker for interstitial fibrosis (41) as it has been extensively validated against the collagen volume fraction (44, 45), it is highly reproducible across different CMR scans (46), it can predict outcomes (47, 48) and histologic validation data show overall the best agreement with ECV compared with other T1 based metrics (46, 49). While native (precontrast) T1 increases and postcontrast T1 decreases with MF, they are inferior MF measures compared with ECV (42, 50). Native T1 reflects changes involving the whole myocardium (intracellular and extracellular compartments) and several confounders affect postcontrast T1 (clearance, body weight, anemia …). Despite this, some studies found a good correlation between native and post-contrast T1 and myocardial fibrosis (51, 52).
Both ECV and T1 are quantitative measures that can allow us to grade disease activity, monitor progress and guide treatment (53). However, the measurements are sensitive to many scanner parameters, with no consensus regarding a vendor-neutral standard approach to image acquisition and postprocessing, so standardization against healthy volunteers is recommended for each CMR scanner and each specific protocol (51, 54). The wide variety of technical and methodological bias-causing confounders makes it difficult to both directly compare native T1 and ECV values between studies.
MF is the final common pathway of a variety of congenital lesions. It is found in both the right and left ventricles and is not limited to areas of surgical scarring or within a coronary distribution (38) (FMF), but it also occurs more diffusely (DMF) in the setting of volume and/or pressure overload (49).
Several studies have characterized MF by CMR in CHD (39, 55); however, there are several limitations. RV disease is hard to assess due to the small thickness of the RV wall, approaching the T1 mapping spatial resolution limit (49). Even in patients with RVs that support systemic circulation, the RV free wall has been deemed difficult to measure by T1 mapping (56) (see Figure 2). Some studies focus on a single mid-ventricular plane, which may not be representative of the entire ventricle, and many CHD patients have limitations for CMR scanning (device electrodes and young age or syndromic problems (38, 57).
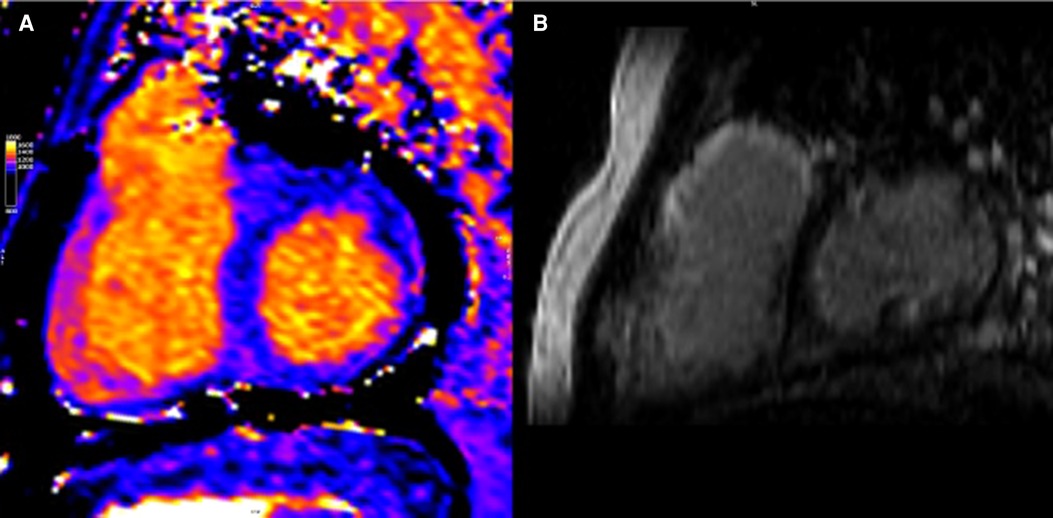
Figure 2. CMR of patient with repaired tetralogy of Fallot. (A) Precontrast T1 mapping of 1,091 ms (normal range of 950–1,050 ms) (B) Extent LGE, especially anterior wall of RVOT and anterior wall of RV.
CHD patients present certain peculiarities and common challenges to CMR when studying MF, such as wide and abnormally shaped QRS complexes, breath-holding difficulties, metallic artifacts from sternal wires and transcatheter devices, as well as pathology focused on the thin-walled RV (39).
Multiple circulating biomarkers of collagen metabolism have been described, but only a few molecules have demonstrated an association between the biomarker levels and histologically assessed myocardial collagen deposition or collagen cross-linking (58–61) (see Figure 3). A positive gradient has been found from the concentration of some of these biomarkers in coronary sinus blood toward the concentration in peripheral vein blood in patients with different types of cardiac disease (58); however, none were cardiac-specific (7).
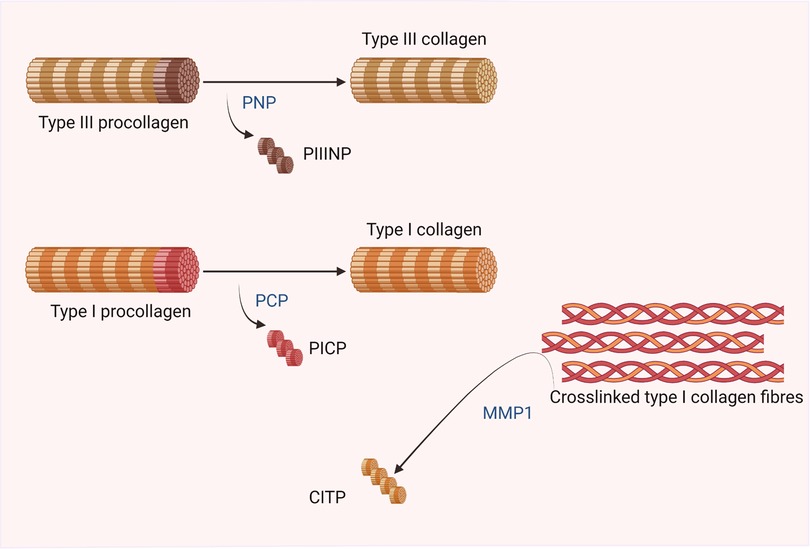
Figure 3. Biomarkers of collagen metabolism. Created with BioRender.com.
The biomarkers that have shown more consistent results and a higher pathological correlation with MF upon biopsy are the carboxy-terminal propeptide of type I procollagen (PICP), the amino-terminal propeptide of type III procollagen (PIIINP) and the ratio of serum carboxy-terminal telopeptide of type I collagen to serum matrix metalloproteinase-1 (CITP:MMP-1ratio) (7, 40). The first two are biomarkers of collagen synthesis that directly correlate with the deposition of collagen fibers in the myocardium in different heart diseases. The CITP:MMP-1 ratio inversely correlates with myocardial collagen cross-linking (59, 62). Therefore, low levels of the CITP:MMP-1 ratio and high levels of PICP and PIIINP would indicate DMF.
PICP is generated during the extracellular maturation of the collagen molecule by conversion of procollagen type I into collagen type I by the enzyme bone morphogenetic protein-1 or procollagen carboxy-terminal proteinase (63).
Serum levels of PICP have been associated with various findings: myocardial deposition of collagen type I fibers in HF patients (23); correlation with severity of HFrEF; with mortality in HFpEF and HFrEF (64, 65) and changes after treatment with torasemide and spironolactone in heart failure (66, 67).
Most serum PIIINP is generated during the extracellular conversion of procollagen type III to collagen type III by the enzyme procollagen amino-terminal proteinase (63). An association has been found between serum PIIINP and myocardial deposition of collagen type III fibers in HF patients and with severity and outcomes in HF of different causes, regardless of the ejection fraction (64, 68).
Once the collagen molecule is mature, the spontaneous self-assembling of molecules results in the formation of collagen fibrils. In a second step, the formation of intra- and intermolecular covalent bonds, cross-links, between lysine residues ensues.
Collagen cross-linking determines the resistance of collagen fibers to MMP degradation; therefore, the highly cross-linked type I collagen fibers have increased resistance to degradation by MMP1, producing a reduction in collagen type I telopeptide (CITP) levels and, thereby, a decreased CITP:MMP1 ratio. Thus, the serum CITP:MMP-1 ratio inversely correlates with myocardial collagen cross-linking (59).
A low CITP:MMP-1 ratio has been associated with a higher risk of hospitalization for HF and cardiovascular mortality in hypertensive patients (59, 69). It also identifies HFpEF patients resistant to the beneficial effects of spironolactone (70).
Myocardial fibrosis is the final common pathway of a variety of CHDs due to the multiple procedures and situations of volume and pressure overload to which these hearts are subjected throughout their lives. The current evidence regarding myocardial fibrosis in specific congenital heart conditions will be reviewed in this chapter, focusing on the most common congenital cardiac disorders.
There is histological evidence of increased interstitial collagen deposition in patients with tetralogy of Fallot (TOF). Existing data suggest that chronic exposure to cyanosis and pressure overload play a role in the development of MF before repair (71). Unloading the RV with a complete repair seems to have a beneficial effect; necropsy studies show a greater extent of MF in unrepaired than repaired TOF (72). However, even after reparative surgery, MF seems to progress in both ventricles and is associated with worse outcomes (72, 73), probably due to residual overloading conditions, usually severe pulmonary regurgitation (see Figure 4).
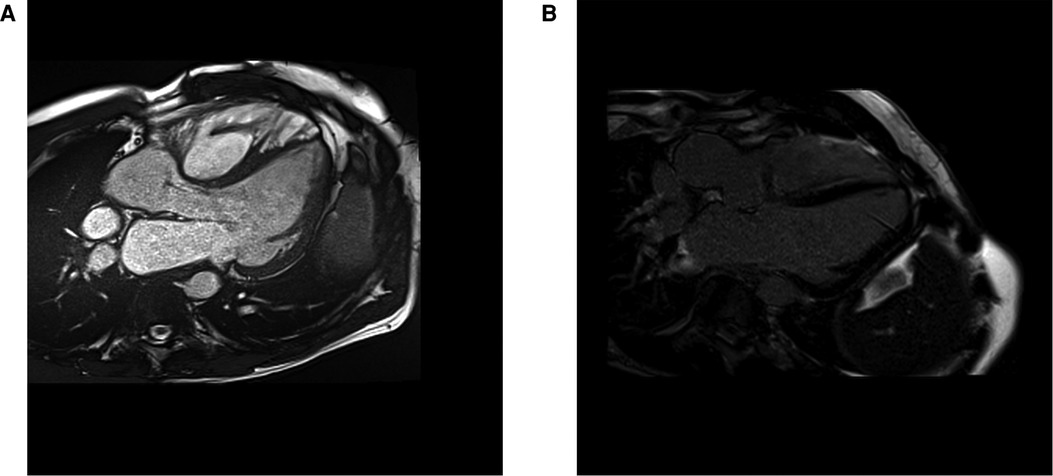
Figure 4. CMR cine 3 chamber view (A) and LGE image (B) from a 39 years old patient with a repaired tetralogy of Fallot, showing focal fibrosis in the anterior wall of the right ventricle.
In 2006, Babu-Narayan et al. (74), published the first study to detect FMF in repaired TOF using LGE. FMF was not only found in areas with patch repair but also distant areas of the RV and even the LV. The extent of LGE was related to age, functional class, neurohormonal activation, ventricular dysfunction and clinical events, including exercise intolerance and clinical arrhythmias. Several studies have since followed, collectively confirming that a greater degree of fibrosis is associated with adverse clinical outcomes (80–82). Additional studies have shown that the presence of fibrosis is not limited to the RV but is also seen in the LV (37), with a positive correlation between LV and RV ECV values, thus indicating various coupling mechanisms and an adverse ventricular-ventricular interaction at the tissue level (73, 75).
RV LGE has been related to RV diastolic dysfunction (83) and fragmentation of the QRS complex, a marker of ventricular arrhythmias in this patient population (84). In a recent study (85), the extent of RV LGE and presence of LV LGE were included in a score that integrated multiple, appropriately weighted, risk factors and which identified the subgroup of patients at the highest annual risk of death.
DMF has also been studied in patients with TOF. Chen and colleagues (73) found a correlation between RV and LV ECVs and an association between predominant volume overloading hemodynamic burden and RV ECV, in contrast with previous evidence supporting pressure overload as the main driver of MF. Since ECV represents the ratio of interstitium relative to total myocardial mass, the authors attributed this finding to a potential myocyte loss as a maladaptative mechanism of cellular remodeling in repaired TOF patients with chronic pulmonary regurgitation. Subsequent studies have also found prolonged RV native T1 times in repaired TOF with volume overload, both in pediatric (86) and adult cohorts (77), which would indicate the presence of DMF. Interestingly, a shorter native T1 was observed in patients with pulmonary valve replacement. This finding, together with histological evidence that patients with repaired TOF and residual pulmonary regurgitation, but reasonably preserved RV volumes and function, show no significant differences in collagen content and LOX activity when compared to healthy controls (87) suggests that DMF might be reversible and even have an oscillating behaviour according to the adaptative capacity of the myocardium to successive overloading conditions.
Elevated RV and LV ECVs have been associated with older age, age at TOF repair, neurohormonal activation, longer QRS duration, shorter six-minute walk distance, larger left atrial volume and adverse outcomes, including death, congestive heart failure and arrhythmias (73, 75, 76).
There are some studies with negative results. Using native T1 mapping, Yigit et al. (88), found focal fibrosis at the RV insertion points and the LV walls in a population of 35 patients with repaired TOF. However, it was not possible to detect RV outflow tract fibrosis with this technique, possibly due to the aforementioned technical limitations of the RV (88).
As for circulating biomarkers, patients with repaired TOF exhibit a pattern of excessive collagen synthesis and dysregulated degradation, with elevated circulating PICP and PIIINP levels and PICP:CITP ratio, a positive correlation between collagen synthesis biomarkers (PICP and PIIINP) and RV end-diastolic volumes (EDV) and a negative correlation with RV systolic function (78, 79).
Atrial switch operation in patients with D-transposition leaves the RV as the systemic pump (89, 90), as it happens naturally in patients born with congenitally corrected transposition of the great arteries (ccTGA) In both situations, a progressive systemic right ventricle (sRV) dilatation and dysfunction are the norm, leading to reduced exercise capacity, HF and arrhythmia (91–93) (see table 2).
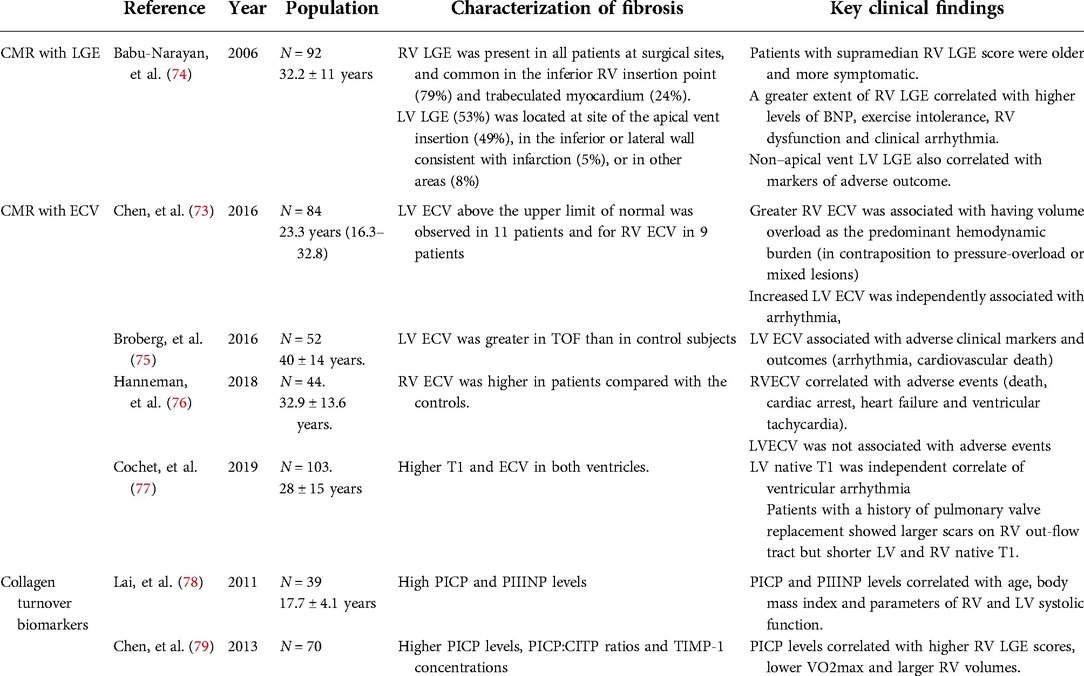
Table 1. Most relevant studies characterizing myocardial interstitial fibrosis with cardiac magnetic resonance and collagen turnover biomarkers in patients with repaired tetralogy of Fallot.
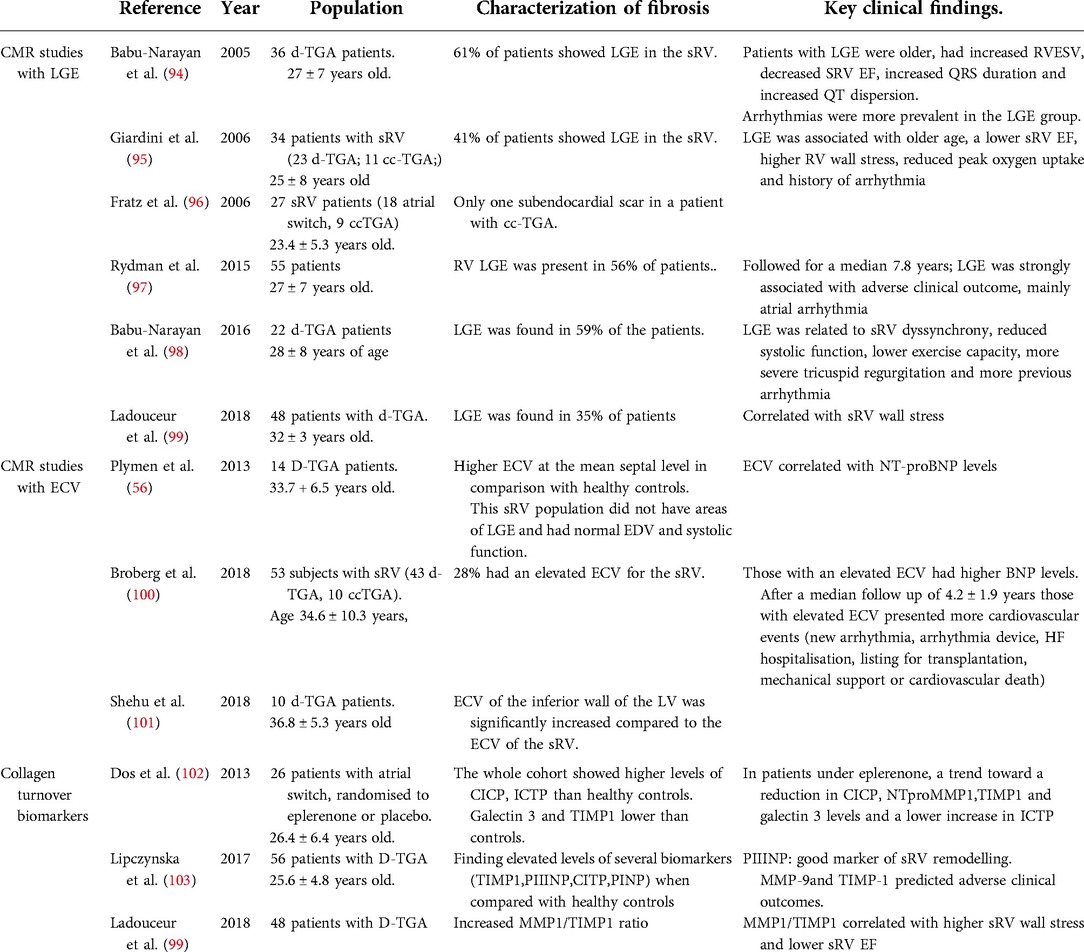
Table 2. Most relevant studies characterizing myocardial interstitial fibrosis with cardiac magnetic resonance and collagen turnover biomarkers in patients with systemic right ventricle.
The first cross-sectional studies characterizing sRV fibrosis via MRI (94–96, 98, 99) showed that the presence of late gadolinium enhancement is common in sRV, ranging from 5% to 60% of patients, probably reflecting methodological differences and heterogenic cohorts. These studies correlated the presence and extent of LGE with functional parameters such as higher sRV end-systolic volume index and lower sRV ejection fraction (EF). Furthermore, the patients more commonly presenting LGE had a prior history of arrhythmia and a lower exercise capacity.
Subsequently, the presence of LGE was validated as a prognostic marker in longitudinal studies. LGE was found to be a good predictor of new onset arrhythmia and heart failure in this population (97).
Lipczynska and colleagues (103) proved that collagen turnover biomarkers were also valuable as prognostic markers in patients with sRV. PIIINP was postulated as a good marker of sRV remodeling, as it correlated with higher sRV mass, higher sRV EDV and worse global longitudinal strain. Furthermore, MMP-9 and tissue inhibitor of metalloproteinase 1(TIMP-1) predicted adverse clinical outcomes in this cohort.
Different mechanisms are postulated to explain late sRV failure and the presence of scarring, which are probably multifactorial. Preoperative hypoxemia and deficient myocardial protection in older cohorts may play a role since more extensive fibrosis has been found in older patients and patients with a late repair (94, 95, 98, 99). A deficient coronary supply for a thickened sRV may be another explanation. Ladouceur and co-workers (99) found that sRV fibrosis was related to increased RV wall stress. Initially, the increased pressure overload for a morphological RV in the systemic position would be compensated by an adaptive hypertrophy to preserve RV function and normalize RV wall stress. However, with time, sRV maladaptation would lead to excessive hypertrophy, which would present demand-supply mismatch ischemia, resulting in the presence of patchy fibrosis and ultimately sRV systolic and diastolic dysfunction. Some studies correlated LGE with QRS duration and QT dispersion (94, 95) and, in the study by Babu-Narayan and colleagues, LGE correlated with echocardiographic parameters of electromechanical delay and dyssynchrony (98), this being another possible mechanism for the deterioration of myocardial function and arrhythmia development.
Recently, CMR studies with T1 mapping techniques for DMF characterization yielded interesting results. Plymen and colleagues (56) found a higher ECV at the mean septal level in a small cohort of 14 patients with sRV in comparison with healthy controls. Interestingly enough, this sRV population did not have areas of LGE and had normal EDV and systolic function (mean RVEDV 79 ml/m2 and mean sRV EF 59%). This could imply that DMF is present in the early stages of the disease and even in the absence of macroscopic scars.
Broberg and co-workers (100) studied 53 subjects with sRV (43 D-TGA, 10 ccTGA);of these, 28% had an elevated ECV value based on gender-specific cut-offs for the systemic LV of healthy controls. No differences were found in sRV volume or EF, but patients with elevated ECV had higher levels of serum brain natriuretic peptide (BNP) and presented more cardiovascular events (new arrhythmia, arrhythmia device, HF hospitalization, listing for transplantation, mechanical support or cardiovascular death)after a median follow up of 4.2 ± 1.9 years. Events in those with elevated ECV tended to occur early and be more related to HF, whereas events in the normal ECV group occurred later and were more often atrial arrhythmias.
More intriguing results focusing on the subpulmonic LV were published recently (101, 104). As in previous studies, a greater native T1 and ECV were found in the sRV, but the authors also found elevated ECV in the subpulmonic LV compared with healthy controls. Moreover, a greater segment and average ECV from the LV was found than in the sRV. The cause of these findings remains poorly understood, although there are several theories, including chronic volume unloading of the eccentrically compressed left ventricle, prevalent postcapillary pulmonary hypertension or ventricular-ventricular interaction at the extracellular matrix level. It also remains unclear whether fibrosis in the subpulmonic LV may play a role in prognosis, but it warrants further research as adverse remodeling with an overt increase in systolic and diastolic volumes of the subpulmonic LV is associated with a worse clinical outcome (105, 106).
The arterial switch operation (ASO) is the current procedure of choice for patients born with D-TGA. In this population, the systemic LV is usually believed to be normal (107–110). However, several factors might impair its myocardial function in the long-term, such as neonatal cyanosis, neonatal cardiopulmonary bypass, impaired coronary perfusion, myocardial denervation, aortic regurgitation or increased aortic stiffness (111) (see Table 3).
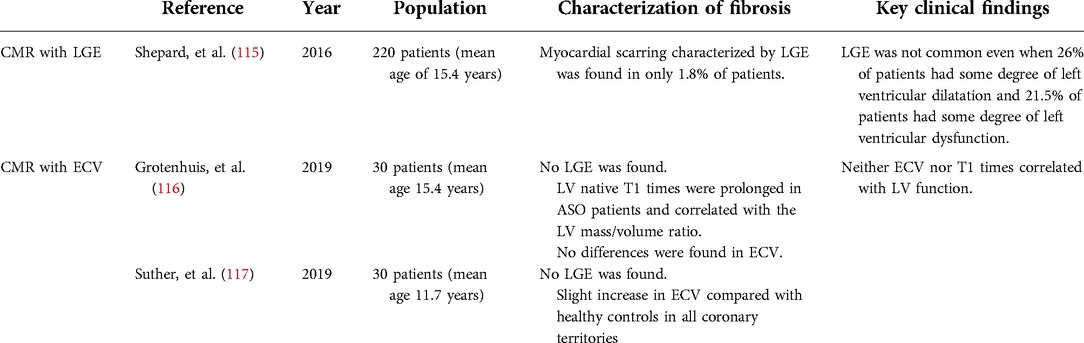
Table 3. Most relevant studies characterizing myocardial fibrosis in patients with arterial switch operation.
Indeed, in the last few years, data with speckle tracking echocardiography have emerged suggesting that a mild impairment of LV function may be more common than previously thought (112–114).
A single-center CMR cohort study (115) in 220 patients (mean age of 15.4 years) found that 26% of patients had some degree of left ventricular dilatation, while 20% had right ventricular dilatation. Left ventricular dysfunction was present in 21.5% of patients (mild in most cases), and only 5.1% of patients had mild right ventricular dysfunction; however, myocardial scarring characterized by LGE was found in only 1.8% of patients.
Diastolic function may also be impaired. Some studies found slight differences in terms of ventricular stiffness and diastolic parameters when compared with healthy controls, but still within the normal range, and in the absence of an established LV hypertrophy (118). This could be explained by an intrinsic stiff myocardium with some degree of fibrotic remodeling, suggested by increased fibrosis markers in CMR studies. Grotenhuis and colleagues (116) studied a cohort of patients without myocardial scarring or perfusion defects and with a normal LV ejection fraction. However, compared with healthy controls, end-diastolic and end-systolic volumes were increased and an altered contraction pattern was observed, the longitudinal strain being lower while the circumferential strain was higher at all short-axis levels, maybe reflecting subclinical compensated systolic dysfunction. LV native T1 times were prolonged in ASO patients and correlated with the LV mass/volume ratio, suggesting the presence of a certain degree of myocardial fibrosis. However, no differences were found in ECV, and neither ECV nor T1 times correlated with LV function. On the other hand, Suther and co-workers detected a slight increase in ECV in a small cohort of adolescents with ASO and normal coronary arteries compared with healthy controls (117). Whether these findings precede the development of overt LV dysfunction over time remains speculative and multicentric longitudinal studies will be required to clarify their prevalence and their prognostic significance.
The assumption that cyanosis causes diffuse intramyocardial fibrosis has been accepted for decades, justified by the findings of fibrosis in specimens from autopsies of patients with unrepaired cyanotic heart disease (see Figure 5 and Table 1). Nevertheless, it remains unclear whether these findings could be related to the presence of adverse hemodynamic loading conditions, the cyanotic condition itself via activation of hypoxia-inducible factors (119, 120) or both.
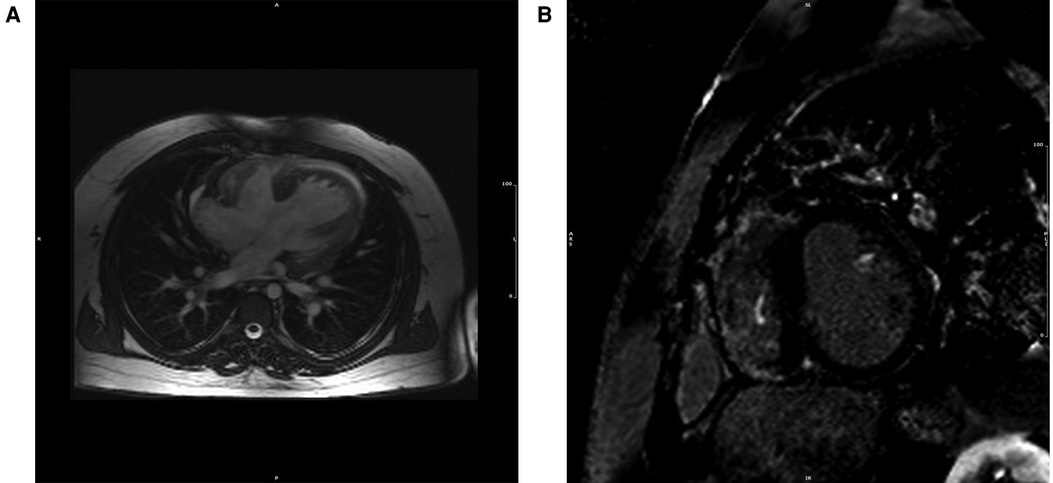
Figure 5. CMR cine (A) and LGE (B) image from 27-year-old patient Eisenmenger syndrome because of an untreated ventricular septal defect, showing extent in right ventricular free wall, as well as in the insertion points and in the left ventricle's papillary muscles. The patient developed ventricular arrhythmias in the follow-up and died suddenly at the age of 33.
In the last few years, this hypothesis has been challenged by some MRI studies mainly focused on patients with Eisenmenger syndrome. However, data are scarce and there are some discrepant results (see Table 4).
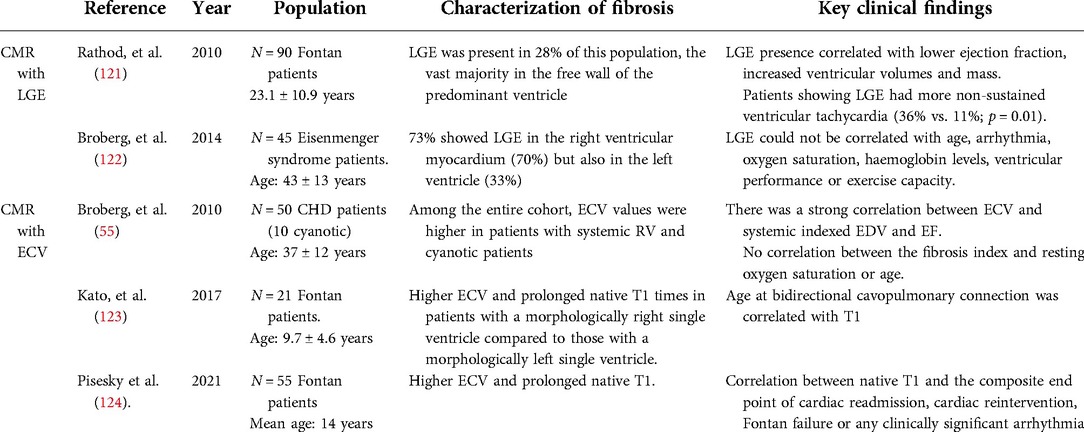
Table 4. Most relevant studies characterizing myocardial fibrosis in patients with chronic cyanosis, Eisenmenger syndrome and Fontan circulation.
Broberg and collaborators (55, 122) quantified ECV in a cohort of 50 CHD patients, 10 of which were cyanotic. Among the entire CHD cohort, cyanotic patients was the group with the second highest ECV, after patients with systemic right ventricle. There was a strong correlation between ECV and systemic indexed EDV and EF. In a larger study from the same group (122) in 45 subjects with Eisenmenger syndrome, LGE was present in 22/30 (73%) patients, specially in the right but also in the left ventricular myocardium. In none of those works fibrosis could be correlated with age, history of arrhythmia, oxygen saturation, hemoglobin levels, exercise capacity or mortality.
On the other hand, Kharabish et al. (125), performed contrast CMR in a very small cohort of patients with repaired and unrepaired cyanotic heart disease. Surprisingly, untreated patients had a significantly lower left ventricular ECV percentage than the repaired patients, and no significant differences were found between cyanotic patients and normal controls.
Both studies were limited by the small cohort size and the heterogeneity of the underlying anatomy, age and physiological state of the patients. Larger multicenter studies are required to unravel the relevance of MF in the pathophysiology of chronic cyanosis and its role in sudden cardiac death and HF, the two leading causes of mortality in this patient population (126).
Myocardial fibrosis has been less studied in this patient population but new techniques for detecting diffuse fibrosis have increased the interest in univentricular patients.
Rathod and colleagues (121) detected FMF in 28% of a population of 90 patients with Fontan circulation. As expected, FMF was found at the surgical sites but the vast majority of LGE distribution was found in the free wall of the predominant ventricle. LGE presence correlated with adverse ventricular mechanics and was associated with non-sustained ventricular tachycardia. Likewise, LGE has been associated with time to death or transplant in this patient population (127).
DMF has also been evaluated in Fontan patients; compared with healthy controls, they presented higher ECV and native T1. Additionally, there was a correlation between native T1 and aortopulmonary collateral flow and an association with the composite end point of cardiac readmission, cardiac reintervention, Fontan failure or any clinically significant arrhythmia (124). However, Kato and colleagues could only find higher ECV in Fontan patients with morphologically right single ventricles. In these cohort, no difference was found regarding T1 or ECV between healthy controls and Fontan patients with morphologically left single ventricles (123).
A recent study using circulating biomarkers in 25 Fontan patients observed a significant correlation between ECV and systemic ventricular end-diastolic pressure and between ECV and liver stiffness. Furthermore, patients with elevated ECV had elevated MMPs and TIMPs, and these patients presented greater liver stiffness (128).
Whether all these findings are related to the intrinsic peculiarities of Fontan circulation, the previous cyanotic phase or both remains to be determined.
As previously mentioned, fibrosis is a highly dynamic process that typically involves the recruitment of fibroblasts and their conversion into myofibroblasts, excessive ECM synthesis and secretion, ECM protein cross-linking, dysregulation of ECM production and breakdown by MMPs and their endogenous inhibitors. Therefore, multiple targets or therapeutic opportunities may exist. In addition, different types of fibrosis (FMF vs. DMF) and different stages of the fibrotic process (more static vs. more dynamic) may coexist in the same individual and identification of the specific or dominant type of fibrosis would be important for the selection of the anti-fibrotic approach. Timely detection of fibrosis and determination of its state could potentially help diagnose and halt HF progression.
There are several potential novel therapeutic strategies for MF coming from the idiopathic pulmonary fibrosis armamentarium (Transforming growth factor beta inhibitor pirfenidone; tyrosine kinase inhibitor nintedanib; pamrevlumab, a human monoclonal antibody that inhibits connective tissue growth factoractivity; simtuzumab, a humanized monoclonal antibody to block lysyl oxidase like 2), some of them showing promising results in preliminary studies (7). However, we still have limited knowledge of the diverse and heterogeneous mechanisms involved in the pathophysiology of the complex process of fibrosis and the different factors (setting, timing or etiology) that might jeopardize the final effectiveness of a given treatment. Also, the therapeutical target should be the excessive fibrous tissue without affecting the physiological collagen scaffold (12); the complexity of this is illustrated by a murine model of myocardial infarction developed by Clarke and colleagues (129). In this experimental model, MMP inhibition proved to be less effective than initially presumed. The authors postulated that early during the remodeling phase, MMP inhibition might be less effective because, although MMP levels are at their highest, there is scant collagen to degrade, while at later fibrotic phases it is less effective because, although collagen concentrations are high, MMP levels have fallen to low levels. On the other hand, MMPs have a full range of functions and myriad targets. All these factors may convey unpredictable and counterintuitive results when targeting the fibrotic process once it has been initiated.
While current experimental studies and even clinical trials [pirfenidone is currently being tested in the PIROUETTE (130) phase II trial] are underway to unravel these riddles, targeting the stimulus that triggered the maladaptative fibrotic process seems to be effective. There is evidence that some HF therapies (targeting neurohormonal activation) have antifibrotic effects (26, 67, 131, 132). However, in CHD, where clinical trials for HF therapy are extremely scarce, those focusing on MF as an endpoint are anecdotal. Our group developed a clinical trial evaluating the effect of the mineralocorticoid-receptor antagonist eplerenone on MF in patients with systemic RV (transposition of the great arteries repaired with the atrial switch procedure) (102). The study found an increased collagen turnover in this patient population compared with age- and gender-matched controls but was underpowered to show a reduction in ventricular mass after one year of treatment. The interpretation of the trend toward a reduction in CICP, MMP1, TIMP1 and galectin 3 levels observed in patients under eplerenone remains elusive as no T1 mapping was available at the time of the study to assess changes in DMF.
While the prognostic role of preoperative myocardial fibrosis in patient outcomes after valvular replacement has been extensively studied (133, 134), there is little information regarding MF reversibility after valvular replacement. However, recent data support the hypothesis DMF may reverse after unloading the ventricles both under volume and pressure overloading conditions (135, 136). The limited data on this topic focuses mainly on mitral and aortic valvular disease with no information, to the best of our knowledge, on right-sided valves. However, experimental studies (artery banding mouse models) suggest that reversibility of RV fibrosis after cessation of the overloading stimulus can also be achieved (137). On the other hand, preliminary and indirect data suggest that DMF in TOF might be reversible and even have an oscillating behavior according to the adaptative capacity of the myocardium to successive overloading conditions (77, 87).
The RV and LV have distinct fibrotic responses and, therefore, may require different therapeutical approaches (138). Pirfenidone does not reverse RV fibrosis or enhance RV function in pulmonary artery-banded rats (139), but it does reduce RV fibrosis and remodeling in a murine model of non-severe pulmonary arterial hypertension (PAH) (140). In turn, eplerenone does not reverse RV fibrosis in Sugen-hypoxia and pulmonary artery-banded mice after PAH was established (141). On the other hand, whereas the microRNA miR-21, a known profibrotic agent currently studied as a target for the treatment of Alport Syndrome, seems to be crucially involved in the fibrotic response to mechanical and hormonal stimuli in isolated RV fibroblasts from dogs, this is not the case for dog LV fibroblasts (19). Whether these discrepant findings are related to the intrinsic different responses of both ventricles to overloading conditions or other determinants of the fibrotic process, such as timing or etiology, remain to be determined.
Wider availability of diagnostic methods to evaluate MF is needed. In recent years, cardiac computed tomography angiography (CTA) has yielded good results for MF assessment. An excellent correlation between ECV evaluated by CTA and by CMR has been shown in animal models, healthy volunteers and HF patients (142, 143). CTA-based quantification of ECV has also been validated against a histological assessment of myocardial fibrosis (142) and presented an association with adverse clinical outcomes after percutaneous aortic valve implant (144, 145). This technique may be of particular interest in patients with CHD since it overcomes some important caveats of CMR in this population: the shorter acquisition times make it easier for patients with cognitive impairment and patients with claustrophobia and it can be used in patients with paramagnetic metal implants.
On the other hand, new targeted approaches that have shown promising results for collagen detection in other organs, such as the novel molecular magnetic resonance contrast agents utilized in liver studies, might be applicable in heart diseases (146).
In summary, understanding HF pathophysiology lies beyond the mere assessment of cardiac function. In-depth consideration of myocardial structure and function at the tissue level, particularly of the myocardial interstitium, provides additional and crucial insight and may drive the development of new therapies. MF, particularly diffuse interstitial fibrosis, constitutes an emerging and promising therapeutic target in HF secondary to acquired heart conditions. Due to intrinsic particularities (preeminent involvement of overloaded morphologically RV, chronic cyanosis, etc.,), MF seems to play a determinant role in the development of HF and arrhythmias in CHD. Further studies in the field are needed to shed light on this exciting and promising area of research in CHD.
All the authors contributed equally to the writing of the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Gross RE, Hubbard JP. Surgical ligation of a patent ductus arteriosus: report of first successful case. JAMA. (1984) 251(9):1201–2. doi: 10.1001/jama.1984.03340330059026
2. Eckhouse SR, Spinale FG. Changes in the myocardial interstitium and contribution to the progression of heart failure. Heart Fail Clin. (2012) 8:7. doi: 10.1016/J.HFC.2011.08.012
3. Weber KT, Jalil JE, Janicki JS, Pick R. Myocardial collagen remodeling in pressure overload hypertrophy. A case for interstitial heart disease. Am J Hypertens. (1989) 2(12 Pt 1):931–40. doi: 10.1093/ajh/2.12.931
4. Collier P, Ledwidge M, McDonald K. Diagnostics and therapeutic interventions in myocardial interstitial disease, a previously neglected pathology. QJM. (2012) 105:721–4. doi: 10.1093/qjmed/hcs072
5. Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation. (1991) 83:1849–65. doi: 10.1161/01.CIR.83.6.1849
6. Vazir A, Fox K, Westaby J, Evans MJ, Westaby S. Can we remove scar and fibrosis from adult human myocardium? Eur Heart J. (2019) 40:960–6. doi: 10.1093/EURHEARTJ/EHY503
7. López B, Ravassa S, Moreno MU, José GS, Beaumont J, González A, et al. Diffuse myocardial fibrosis: mechanisms, diagnosis and therapeutic approaches. Nat Rev Cardiol. (2021) 18:479–98. doi: 10.1038/S41569-020-00504-1
8. Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, et al. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife. (2019) 8:e43882. doi: 10.7554/ELIFE.43882
9. Lijnen P, Petrov V. Induction of cardiac fibrosis by aldosterone. J Mol Cell Cardiol. (2000) 32:865–79. doi: 10.1006/JMCC.2000.1129
10. Collier P, Watson CJ, van Es MH, Phelan D, McGorrian C, Tolan M, et al. Getting to the heart of cardiac remodeling; how collagen subtypes may contribute to phenotype. J Mol Cell Cardiol. (2012) 52:148–53. doi: 10.1016/J.YJMCC.2011.10.002
11. Schelbert EB, Butler J, Diez J. Why clinicians should care about the cardiac interstitium. JACC Cardiovasc Imaging. (2019) 12:2305–18. doi: 10.1016/J.JCMG.2019.04.025
12. de Boer RA, de Keulenaer G, Bauersachs J, Brutsaert D, Cleland JG, Diez J, et al. Towards better definition, quantification and treatment of fibrosis in heart failure. A scientific roadmap by the Committee of Translational Research of the Heart Failure Association (HFA) of the European Society of Cardiology. Eur J Heart Fail. (2019) 21:272–85. doi: 10.1002/EJHF.1406
13. Beltrami CA, Finato N, Rocco M, Feruglio GA, Puricelli C, Cigola E, et al. Structural basis of end-stage failure in ischemic cardiomyopathy in humans. Circulation. (1994) 89:151–63. doi: 10.1161/01.CIR.89.1.151
14. Hartupee J, Mann DL. Neurohormonal activation in heart failure with reduced ejection fraction. Nat Rev Cardiol. (2017) 14:30–8. doi: 10.1038/NRCARDIO.2016.163
15. Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. (2013) 62:263–71. doi: 10.1016/J.JACC.2013.02.092
16. Friedberg MK, Redington AN. Right versus left ventricular failure: differences, similarities, and interactions. Circulation. (2014) 129:1033–44. doi: 10.1161/CIRCULATIONAHA.113.001375
17. Oken DE, Boucek RJ. Quantitation of collagen in human myocardium. Circ Res. (1957) 5:357–61. doi: 10.1161/01.RES.5.4.357
18. Herpel E, Singer S, Flechtenmacher C, Pritsch M, Sack FU, Hagl S, et al. Extracellular matrix proteins and matrix metalloproteinases differ between various right and left ventricular sites in end-stage cardiomyopathies. Virchows Arch. (2005) 446:369–78. doi: 10.1007/S00428-004-1177-Z
19. Powers JC, Sabri A, Al-Bataineh D, Chotalia D, Guo X, Tsipenyuk F, et al. Differential microRNA-21 and microRNA-221 upregulation in the biventricular failing heart reveals distinct stress responses of right versus left ventricular fibroblasts. Circ Heart Fail. (2020) 13:e006426. doi: 10.1161/CIRCHEARTFAILURE.119.006426
20. Bernal-Ramirez J, Diaz-Vesga MC, Talamilla M, Mendez A, Quiroga C, Garza-Cervantes JA, et al. Exploring functional differences between the right and left ventricles to better understand right ventricular dysfunction. Oxid Med Cell Longev. (2021) 2021:9993060. doi: 10.1155/2021/9993060
21. Robinson TF, Factor SM, Sonnenblick EH. The heart as a suction pump. Sci Am. (1986) 254(6):84–91. doi: 10.1038/scientificamerican0686-84
22. Echegaray K, Andreu I, Lazkano A, Villanueva I, Sáenz A, Elizalde MR, et al. Role of myocardial collagen in severe aortic stenosis with preserved ejection fraction and symptoms of heart failure. Rev Esp Cardiol (Engl Ed). (2017) 70:832–40. doi: 10.1016/j.rec.2016.12.038
23. López B, Querejeta R, González A, Larman M, Díez J. Collagen cross-linking but not collagen amount associates with elevated filling pressures in hypertensive patients with stage C heart failure potential role of lysyl oxidase. Hypertension. (2012) 60(3):677–83. doi: 10.1161/HYPERTENSIONAHA.112.196113
24. Kasner M, Westermann D, Lopez B, Gaub R, Escher F, Kühl U, et al. Diastolic tissue Doppler indexes correlate with the degree of collagen expression and cross-linking in heart failure and normal ejection fraction. J Am Coll Cardiol. (2011) 57:977–85. doi: 10.1016/j.jacc.2010.10.024
25. Querejeta R, López B, González A, Sánchez E, Larman M, Martínez Ubago JL, et al. Increased collagen type I synthesis in patients with heart failure of hypertensive origin: relation to myocardial fibrosis. Circulation. (2004) 110:1263–8. doi: 10.1161/01.CIR.0000140973.60992.9A
26. Díez J, Querejeta R, López B, González A, Larman M, Martínez Ubago JL. Losartan-dependent regression of myocardial fibrosis is associated with reduction of left ventricular chamber stiffness in hypertensive patients. Circulation. (2002) 105:2512–7. doi: 10.1161/01.CIR.0000017264.66561.3D
27. González A, López B, Querejeta R, Zubillaga E, Echeverría T, Díez J. Filling pressures and collagen metabolism in hypertensive patients with heart failure and normal ejection fraction. Hypertension. (2010) 55:1418–24. doi: 10.1161/HYPERTENSIONAHA.109.149112
28. López B, González A, Querejeta R, Larman M, Díez J. Alterations in the pattern of collagen deposition may contribute to the deterioration of systolic function in hypertensive patients with heart failure. J Am Coll Cardiol. (2006) 48:89–96. doi: 10.1016/j.jacc.2006.01.077
29. Baicu CF, Stroud JD, Livesay VA, Hapke E, Holder J, Spinale FG, et al. Changes in extracellular collagen matrix alter myocardial systolic performance. Am J Physiol Heart Circ Physiol. (2003) 284(1):H122–32. doi: 10.1152/ajpheart.00233.2002
30. Villari B, Campbell SE, Hess OM, Mall G, Vassalli G, Weber KT, et al. Influence of collagen network on left ventricular systolic and diastolic function in aortic valve disease. J Am Coll Cardiol. (1993) 22:1477–84. doi: 10.1016/0735-1097(93)90560-N
31. André A, Kléber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev. (2004) 84(2):431–88. doi: 10.1152/physrev.00025.2003
32. Nguyen MN, Kiriazis H, Gao XM, Du XJ. Cardiac fibrosis and arrhythmogenesis. Compr Physiol. (2017) 7:1009–49. doi: 10.1002/cphy.c160046
33. He X, Gao X, Peng L, Wang S, Zhu Y, Ma H, et al. Atrial fibrillation induces myocardial fibrosis through angiotensin II type 1 receptor-specific Arkadia-mediated downregulation of Smad7. Circ Res. (2011) 108:164–75. doi: 10.1161/CIRCRESAHA.110.234369
34. Tsai CT, den Tseng C, Hwang JJ, Wu CK, Yu CC, Wang YC, et al. Tachycardia of atrial myocytes induces collagen expression in atrial fibroblasts through transforming growth factor β1. Cardiovasc Res. (2011) 89:805–15. doi: 10.1093/cvr/cvq322
35. He X, Zhang K, Gao X, Li L, Tan H, Chen J, et al. Rapid atrial pacing induces myocardial fibrosis by down-regulating Smad7 via microRNA-21 in rabbit. Heart Vessels. (2016) 31:1696–708. doi: 10.1007/s00380-016-0808-z
36. González A, Schelbert EB, Díez J, Butler J. Myocardial interstitial fibrosis in heart failure: biological and translational perspectives. J Am Coll Cardiol. (2018) 71:1696–706. doi: 10.1016/j.jacc.2018.02.021
37. Broberg CS, Khan AM. Myocardial fibrosis in adult congenital heart disease. Rev Esp Cardiol. (2020) 73:707–10. doi: 10.1016/j.recesp.2020.02.014
38. Broberg CS, Burchill LJ. Myocardial factor revisited: the importance of myocardial fibrosis in adults with congenital heart disease. Int J Cardiol. (2015) 189:204–10. doi: 10.1016/j.ijcard.2015.04.064
39. Ghonim S, Voges I, Gatehouse PD, Keegan J, Gatzoulis MA, Kilner PJ, et al. Myocardial architecture, mechanics, and fibrosis in congenital heart disease. Front Cardiovasc Med. (2017) 4:30. doi: 10.3389/fcvm.2017.00030
40. Gyöngyösi M, Winkler J, Ramos I, Do QT, Firat H, McDonald K, et al. Myocardial fibrosis: biomedical research from bench to bedside. Eur J Heart Fail. (2017) 19:177–91. doi: 10.1002/ejhf.696
41. Rathod RH, Powell AJ, Geva T. Myocardial fibrosis in congenital heart disease. Circ J. (2016) 80:1300–7. doi: 10.1253/circj.CJ-16-0353
42. Schelbert EB, Sabbah HN, Butler J, Gheorghiade M. Employing extracellular volume cardiovascular magnetic resonance measures of myocardial fibrosis to foster novel therapeutics. Circ Cardiovasc Imaging. (2017) 10(6):e005619. doi: 10.1161/CIRCIMAGING.116.005619
43. Robinson AA, Chow K, Salerno M. Myocardial T1 and ECV measurement: underlying concepts and technical considerations. JACC Cardiovasc Imaging. (2019) 12:2332–44. doi: 10.1016/j.jcmg.2019.06.031
44. de Meester De Ravenstein C, Bouzin C, Lazam S, Boulif J, Amzulescu M, Melchior J, et al. Histological validation of measurement of diffuse interstitial myocardial fibrosis by myocardial extravascular volume fraction from Modified Look-Locker imaging (MOLLI) T1 mapping at 3 T. J Cardiovasc Magn Reson. (2015) 17:48. doi: 10.1186/s12968-015-0150-0
45. Miller CA, Naish JH, Bishop P, Coutts G, Clark D, Zhao S, et al. Comprehensive validation of cardiovascular magnetic resonance techniques for the assessment of myocardial extracellular volume. Circ Cardiovasc Imaging. (2013) 6:373–83. doi: 10.1161/CIRCIMAGING.112.000192
46. Fontana M, White SK, Banypersad SM, Sado DM, Maestrini V, Flett AS, et al. Comparison of T1 mapping techniques for ECV quantification. Histological validation and reproducibility of ShMOLLI versus multibreath-hold T1 quantification equilibrium contrast CMR. J Cardiovasc Magn Reson. (2012) 14:88. doi: 10.1186/1532-429X-14-88
47. Zhuang B, Sirajuddin A, Wang S, Arai A, Zhao S, Lu M. Prognostic value of T1 mapping and extracellular volume fraction in cardiovascular disease: a systematic review and meta-analysis. Heart Fail Rev. (2018) 23:723–31. doi: 10.1007/s10741-018-9718-8
48. Duca F, Zotter-Tufaro C, Kammerlander AA, Panzenböck A, Aschauer S, Dalos D, et al. Cardiac extracellular matrix is associated with adverse outcome in patients with chronic heart failure. Eur J Heart Fail. (2017) 19:502–11. doi: 10.1002/ejhf.680
49. Messroghli DR, Moon JC, Ferreira VM, Grosse-Wortmann L, He T, Kellman P, et al. Clinical recommendations for cardiovascular magnetic resonance mapping of T1, T2, T2 and extracellular volume: a consensus statement by the Society for Cardiovascular Magnetic Resonance (SCMR) endorsed by the European Association for Cardiovascular Imaging (EACVI). J Cardiovasc Magn Reson. (2017) 19(1):75. doi: 10.1186/s12968-017-0389-8
50. Inui K, Tachi M, Saito T, Kubota Y, Murai K, Kato K, et al. Superiority of the extracellular volume fraction over the myocardial T1 value for the assessment of myocardial fibrosis in patients with non-ischemic cardiomyopathy. Magn Reson Imaging. (2016) 34:1141–5. doi: 10.1016/j.mri.2016.05.008
51. Diao K-Y, Yang Z-G, Xu H-Y, Liu X, Zhang Q, Shi K, et al. Histologic validation of myocardial fibrosis measured by T1 mapping: a systematic review and meta-analysis. J Cardiovasc Magn Reson. (2016) 18:1–11. doi: 10.1186/s12968-016-0313-7
52. Puntmann VO, Carr-White G, Jabbour A, Yu CY, Gebker R, Kelle S, et al. T1-mapping and outcome in nonischemic cardiomyopathy all-cause mortality and heart failure. JACC Cardiovasc Imaging. (2016) 9:40–50. doi: 10.1016/j.jcmg.2015.12.001
53. Puntmann VO, Peker E, Chandrashekhar Y, Nagel E. T1 mapping in characterizing myocardial disease: a comprehensive review. Circ Res. (2016) 119:277–99. doi: 10.1161/CIRCRESAHA.116.307974
54. Moon JC, Messroghli DR, Kellman P, Piechnik SK, Robson MD, Ugander M, et al. Myocardial T1 mapping and extracellular volume quantification: a Society for Cardiovascular Magnetic Resonance (SCMR) and CMR Working Group of the European Society of Cardiology consensus statement. J Cardiovasc Magn Reson. (2013) 15:1–12. doi: 10.1186/1532-429X-15-92
55. Broberg CS, Chugh SS, Conklin C, Sahn DJ, Jerosch-Herold M. Quantification of diffuse myocardial fibrosis and its association with myocardial dysfunction in congenital heart disease. Circ Cardiovasc Imaging. (2010) 3:727–34. doi: 10.1161/CIRCIMAGING.108.842096
56. Plymen CM, Sado DM, Taylor AM, Bolger AP, Lambiase PD, Hughes M, et al. Diffuse myocardial fibrosis in the systemic right ventricle of patients late after Mustard or Senning surgery: an equilibrium contrast cardiovascular magnetic resonance study. Eur Heart J Cardiovasc Imaging. (2013) 14:963–8. doi: 10.1093/ehjci/jet014
57. Riesenkampff E, Messroghli DR, Redington AN, Grosse-Wortmann L. Myocardial T1 mapping in pediatric and congenital heart disease. Circ Cardiovasc Imaging. (2015) 8(2):e002504. doi: 10.1161/CIRCIMAGING.114.002504
58. López B, González A, Díez J. Circulating biomarkers of collagen metabolism in cardiac diseases. Circulation. (2010) 121:1645–54. doi: 10.1161/CIRCULATIONAHA.109.912774
59. López B, Ravassa S, González A, Zubillaga E, Bonavila C, Bergés M, et al. Myocardial collagen cross-linking is associated with heart failure hospitalization in patients with hypertensive heart failure. J Am Coll Cardiol. (2016) 67:251–60. doi: 10.1016/j.jacc.2015.10.063
60. Chalikias GK, Tziakas DN. Biomarkers of the extracellular matrix and of collagen fragments. Clin Chim Acta. (2015) 443:39–47. doi: 10.1016/j.cca.2014.06.028
61. Ureche C, Nedelcu AE, Sascau RA, Statescu C, Kanbay M, Covic A. Role of collagen turnover biomarkers in the noninvasive assessment of myocardial fibrosis: an update. Biomark Med. (2020) 14:1265–75. doi: 10.2217/bmm-2020-0298
62. Klappacher G, Franzen P, Haab D, Mehrabi M, Binder M, Plesch K, et al. Measuring Extracellular Matrix Turnover in the Serum of Patients With Idiopathic or lschemic Dilated Cardiomyopathy and Impact on Diagnosis and Prognosis. Am J Cardiol. (1995) 75(14):913–8. doi: 10.1016/s0002-9149(99)80686-9
63. Prockop DJ, Kivirikko KI. COLLAGENS: Molecular Biology, Diseases, and Potentials for Therapy. (1995). www.annualreviews.org
64. Krum H, Elsik M, Schneider HG, Ptaszynska A, Black M, Carson PE, et al. Relation of peripheral collagen markers to death and hospitalization in patients with heart failure and preserved ejection fraction results of the I-PRESERVE collagen substudy. Circ Heart Fail. (2011) 4:561–8. doi: 10.1161/CIRCHEARTFAILURE.110.960716
65. Löfsjögård J, Kahan T, Díez J, López B, González A, Ravassa S, et al. Usefulness of collagen carboxy-terminal propeptide and telopeptide to predict disturbances of long-term mortality in patients ≥60 years with heart failure and reduced ejection fraction. Am J Cardiol. (2017) 119:2042–8. doi: 10.1016/j.amjcard.2017.03.036
66. López B, Querejeta R, González A, Sánchez E, Larman M, Díez J. Effects of loop diuretics on myocardial fibrosis and collagen type I turnover in chronic heart failure. J Am Coll Cardiol. (2004) 43:2028–35. doi: 10.1016/j.jacc.2003.12.052
67. Izawa H, Murohara T, Nagata K, Isobe S, Asano H, Amano T, et al. Mineralocorticoid receptor antagonism ameliorates left ventricular diastolic dysfunction and myocardial fibrosis in mildly symptomatic patients with idiopathic dilated cardiomyopathy: a pilot study. Circulation. (2005) 112:2940–5. doi: 10.1161/CIRCULATIONAHA.105.571653
68. Zannad F, Alla F, Dousset B, Perez A, Pitt B. Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure insights from the randomized aldactone evaluation study (RALES). Circulation. (2000) 102(22):2700–6. doi: 10.1161/01.cir.102.22.270011094035
69. Ravassaa S, López B, Querejeta R, Echegaray K, José GS, Moreno MU, et al. Phenotyping of myocardial fibrosis in hypertensive patients with heart failure. Influence on clinical outcome. J Hypertens. (2017) 35:853–61. doi: 10.1097/HJH.0000000000001258
70. Ravassa S, Trippel T, Bach D, Bachran D, González A, López B, et al. Biomarker-based phenotyping of myocardial fibrosis identifies patients with heart failure with preserved ejection fraction resistant to the beneficial effects of spironolactone: results from the Aldo-DHF trial. Eur J Heart Fail. (2018) 20:1290–9. doi: 10.1002/ejhf.1194
71. Chowdhury UK, Sathia S, Ray R, Singh R, Pradeep KK, Venugopal P. Histopathology of the right ventricular outflow tract and its relationship to clinical outcomes and arrhythmias in patients with tetralogy of Fallot. J Thorac Cardiovasc Surg. (2006) 132(2):270–7. doi: 10.1016/J.JTCVS.2006.04.001
72. Pradegan N, Vida VL, Geva T, Stellin G, White MT, Sanders SP, et al. Myocardial histopathology in late-repaired and unrepaired adults with tetralogy of Fallot. Cardiovasc Pathol. (2016) 25:225–31. doi: 10.1016/J.CARPATH.2016.02.001
73. Chen CA, Dusenbery SM, Valente AM, Powell AJ, Geva T. Myocardial ECV fraction assessed by CMR is associated with type of hemodynamic load and arrhythmia in repaired tetralogy of Fallot. JACC Cardiovasc Imaging. (2016) 9:1–10. doi: 10.1016/j.jcmg.2015.09.011
74. Babu-Narayan SV, Kilner PJ, Li W, Moon JC, Goktekin O, Davlouros PA, et al. Ventricular fibrosis suggested by cardiovascular magnetic resonance in adults with repaired tetralogy of Fallot and its relationship to adverse markers of clinical outcome. Circulation. (2006) 113:405–13. doi: 10.1161/CIRCULATIONAHA.105.548727
75. Broberg CS, Huang J, Hogberg I, McLarry J, Woods P, Burchill LJ, et al. Diffuse LV myocardial fibrosis and its clinical associations in adults with repaired tetralogy of Fallot. JACC Cardiovasc Imaging. (2016) 9:86–7. doi: 10.1016/j.jcmg.2015.10.006
76. Hanneman K, Crean AM, Wintersperger BJ, Thavendiranathan P, Nguyen ET, Kayedpour C, et al. The relationship between cardiovascular magnetic resonance imaging measurement of extracellular volume fraction and clinical outcomes in adults with repaired tetralogy of Fallot. Eur Heart J Cardiovasc Imaging. (2018) 19:777–84. doi: 10.1093/ehjci/jex248
77. Cochet H, Iriart X, Allain-Nicolaï A, Camaioni C, Sridi S, Nivet H, et al. Focal scar and diffuse myocardial fibrosis are independent imaging markers in repaired tetralogy of Fallot. Eur Heart J Cardiovasc Imaging. (2019) 20:990–1003. doi: 10.1093/ehjci/jez068
78. Lai CTM, Chan KW, Wong SJ, Chow PC, Cheung YF. Circulating levels of biomarkers of collagen synthesis and ventricular function and dyssynchrony in adolescents and young adults after repair of tetralogy of Fallot. Am Heart J. (2011) 162:467–73. doi: 10.1016/J.AHJ.2011.05.027
79. Chen CA, Tseng WYI, Wang JK, Chen SY, Ni YH, Huang KC, et al. Circulating biomarkers of collagen type i metabolism mark the right ventricular fibrosis and adverse markers of clinical outcome in adults with repaired tetralogy of Fallot. Int J Cardiol. (2013) 167:2963–8. doi: 10.1016/j.ijcard.2012.08.059
80. Ylitalo P, Pitkänen OM, Lauerma K, Holmström M, Rahkonen O, Heikinheimo M, et al. Late gadolinium enhancement (LGE) progresses with right ventricle volume in children after repair of tetralogy of Fallot. IJC Heart Vessels. (2014) 3:15–20. doi: 10.1016/j.ijchv.2014.01.002
81. Sṕiewak M, Małek ŁA, Petryka J, Mazurkiewicz Ł, Marczak M, Biernacka EK, et al. Factors associated with biventricular dysfunction in patients with repaired tetralogy of Fallot. Kardiol Pol. (2014) 72:631–9. doi: 10.5603/KP.a2014.0049
82. Dobson RJ, Mordi I, Danton MH, Walker NL, Walker HA, Tzemos N. Late gadolinium enhancement and adverse outcomes in a contemporary cohort of adult survivors of tetralogy of Fallot. Congenit Heart Dis. (2017) 12:58–66. doi: 10.1111/chd.12403
83. Munkhammar P, Carlsson M, Arheden H, Pesonen E. Restrictive right ventricular physiology after Tetralogy of Fallot repair is associated with fibrosis of the right ventricular outflow tract visualized on cardiac magnetic resonance imaging. Eur Heart J Cardiovasc Imaging. (2013) 14:978–85. doi: 10.1093/ehjci/jet009
84. Park SJ, On YK, Kim JS, Park SW, Yang JH, Jun TG, et al. Relation of fragmented QRS complex to right ventricular fibrosis detected by late gadolinium enhancement cardiac magnetic resonance in adults with repaired tetralogy of Fallot. Am J Cardiol. (2012) 109:110–5. doi: 10.1016/j.amjcard.2011.07.070
85. Ghonim S, Gatzoulis MA, Ernst S, Li W, Moon JC, Smith GC, et al. Predicting survival in repaired tetralogy of Fallot. JACC Cardiovasc Imaging. (2022) 15(2):257–68. doi: 10.1016/j.jcmg.2021.07.026
86. Yim D, Riesenkampff E, Caro-Dominguez P, Yoo SJ, Seed M, Grosse-Wortmann L. Assessment of diffuse ventricular myocardial fibrosis using native T1 in children with repaired tetralogy of Fallot. Circ Cardiovasc Imaging. (2017) 10(3):e005695. doi: 10.1161/CIRCIMAGING.116.005695
87. Brayson D, Holohan SJ, Bardswell SC, Arno M, Lu H, Jensen HK, et al. Right ventricle has Normal myofilament function but shows perturbations in the expression of extracellular matrix genes in patients with tetralogy of Fallot undergoing pulmonary valve replacement. J Am Heart Assoc. (2020) 9(16):e015342. doi: 10.1161/JAHA.119.015342
88. Yiğit H, Ergün E, Öztekin PS, Koşar PN. Can T1 mapping be an alternative of post-contrast magnetic resonance sequences in patients with surgically corrected tetralogy of Fallot? Anatol J Cardiol. (2020) 24:377–81. doi: 10.14744/AnatolJCardiol.2020.73576
89. Senning Å. Surgical correction of transposition of the great vessels. Surgery. (1959) 45:966–80.13659340
90. Mustard W. Successful two-stage correction of transposition of the great vessels. Surgery. (1964) 55:469–72.14133108
91. Piran S, Veldtman G, Siu S, Webb GD, Liu PP. Heart failure and ventricular dysfunction in patients with single or systemic right ventricles. Circulation. (2002) 105:1189–94. doi: 10.1161/hc1002.105182
92. Gatzoulis MA, Walters J, McLaughlin PR, Merchant N, Webb GD, Liu P. Late arrhythmia in adults with the Mustard procedure for transposition of great arteries: a surrogate marker for right ventricular dysfunction? Heart. (2000) 84:409–15. doi: 10.1136/heart.84.4.409
93. Couperus LE, Vliegen HW, Zandstra TE, Kiès P, Jongbloed MRM, Holman ER, et al. Long-term outcome after atrial correction for transposition of the great arteries. Heart. (2019) 105:790–6. doi: 10.1136/heartjnl-2018-313647
94. Babu-Narayan SV, Goktekin O, Moon JC, Broberg CS, Pantely GA, Pennell DJ, et al. Late gadolinium enhancement cardiovascular magnetic resonance of the systemic right ventricle in adults with previous atrial redirection surgery for transposition of the great arteries. Circulation. (2005) 111:2091–8. doi: 10.1161/01.CIR.0000162463.61626.3B
95. Giardini A, Lovato L, Donti A, Formigari R, Oppido G, Gargiulo G, et al. Relation between right ventricular structural alterations and markers of adverse clinical outcome in adults with systemic right ventricle and either congenital complete (after senning operation) or congenitally corrected transposition of the great arteries. Am J Cardiol. (2006) 98:1277–82. doi: 10.1016/j.amjcard.2006.05.062
96. Fratz S, Hauser M, Bengel FM, Hager A, Kaemmerer H, Schwaiger M, et al. Myocardial scars determined by delayed-enhancement magnetic resonance imaging and positron emission tomography are not common in right ventricles with systemic function in long-term follow up. Heart. (2006) 92:1673–7. doi: 10.1136/hrt.2005.086579
97. Rydman R, Gatzoulis MA, Ho SY, Ernst S, Swan L, Li W, et al. Systemic right ventricular fibrosis detected by cardiovascular magnetic resonance is associated with clinical outcome, mainly new-onset atrial arrhythmia, in patients after atrial redirection surgery for transposition of the great arteries. Circ Cardiovasc Imaging. (2015) 8(5):e002628. doi: 10.1161/CIRCIMAGING.114.002628
98. Babu-Narayan SV, Prati D, Rydman R, Dimopoulos K, Diller GP, Uebing A, et al. Dyssynchrony and electromechanical delay are associated with focal fibrosis in the systemic right ventricle — insights from echocardiography. Int J Cardiol. (2016) 220:382–8. doi: 10.1016/j.ijcard.2016.06.090
99. Ladouceur M, Baron S, Nivet-Antoine V, Maruani G, Soulat G, Pereira H, et al. Role of myocardial collagen degradation and fibrosis in right ventricle dysfunction in transposition of the great arteries after atrial switch. Int J Cardiol. (2018) 258:76–82. doi: 10.1016/j.ijcard.2018.01.100
100. Broberg CS, Valente AM, Huang J, Burchill LJ, Holt J, van Woerkom R, et al. Myocardial fibrosis and its relation to adverse outcome in transposition of the great arteries with a systemic right ventricle. Int J Cardiol. (2018) 271:60–5. doi: 10.1016/j.ijcard.2018.04.089
101. Shehu N, Meierhofer C, Messroghli D, Mkrtchyan N, Martinoff S, Ewert P, et al. Diffuse fibrosis is common in the left, but not in the right ventricle in patients with transposition of the great arteries late after atrial switch operation. Int J Cardiovasc Imaging. (2018) 34:1241–8. doi: 10.1007/s10554-018-1338-9
102. Dos L, Pujadas S, Estruch M, Mas A, Ferreira-González I, Pijuan A, et al. Eplerenone in systemic right ventricle: double blind randomized clinical trial. The evedes study. Int J Cardiol. (2013) 168:5167–73. doi: 10.1016/j.ijcard.2013.07.163
103. Lipczyńska M, Szymański P, Kumor M, Klisiewicz A, Hoffman P. Collagen turnover biomarkers and systemic right ventricle remodeling in adults with previous atrial switch procedure for transposition of the great arteries. PLoS ONE. (2017) 12(8):e0180629. doi: 10.1371/journal.pone.0180629
104. Chen RHS, Wong SJ, Wong WHS, Cheung YF. Left ventricular contractile reserve after arterial switch operation for complete transposition of the great arteries: an exercise echocardiographic study. Eur Heart J Cardiovasc Imaging. (2013) 14:480–6. doi: 10.1093/ehjci/jes204
105. Surkova E, Segura T, Dimopoulos K, Bispo D, Flick C, West C, et al. Systolic dysfunction of the subpulmonary left ventricle is associated with the severity of heart failure in patients with a systemic right ventricle. Int J Cardiol. (2021) 324:66–71. doi: 10.1016/j.ijcard.2020.09.051
106. Santens B, Helsen F, van de Bruaene A, de Meester P, Budts A-L, Troost E, et al. Adverse functional remodelling of the subpulmonary left ventricle in patients with a systemic right ventricle is associated with clinical outcome. Eur Heart J Cardiovasc Imaging. (2022) 23:680–8. doi: 10.1093/ehjci/jeab086
107. Fricke TA, Buratto E, Weintraub RG, Bullock A, Wheaton G, Grigg L, et al. Long-term outcomes of the arterial switch operation. J Thorac Cardiovasc Surg. (2022) 163:212–9. doi: 10.1016/j.jtcvs.2021.01.134
108. van der Palen RLF, Blom NA, Kuipers IM, Rammeloo LAJ, Jongbloed MRM, Konings TC, et al. Long-term outcome after the arterial switch operation: 43 years of experience. Eur J Cardiothorac Surg. (2021) 59:968–77. doi: 10.1093/ejcts/ezab006
109. Santens B, van de Bruaene A, de Meester P, Gewillig M, Troost E, Claus P, et al. Outcome of arterial switch operation for transposition of the great arteries. A 35-year follow-up study. Int J Cardiol. (2020) 316:94–100. doi: 10.1016/j.ijcard.2020.04.072
110. van Wijk SW, Driessen MM, Meijboom FJ, Doevendans PA, Schoof PH, Breur HM, et al. Left ventricular function and exercise capacity after arterial switch operation for transposition of the great arteries: a systematic review and meta-analysis. Cardiol Young. (2018) 28:895–902. doi: 10.1017/S1047951117001032
111. Biglino G, Ntsinjana H, Plymen C, Tann O, Giardini A, Derrick G, et al. Ventriculovascular interactions late after atrial and arterial repair of transposition of the great arteries. J Thorac Cardiovasc Surg. (2014) 148:2627–33. doi: 10.1016/j.jtcvs.2014.07.072
112. Pettersen E, Fredriksen PM, Urheim S, Thaulow E, Smith HJ, Smevik B, et al. Ventricular function in patients with transposition of the great arteries operated with arterial switch. Am J Cardiol. (2009) 104:583–9. doi: 10.1016/j.amjcard.2009.04.029
113. van Wijk SW, Driessen MMP, Meijboom FJ, Takken T, Doevendans PA, Breur JM. Evaluation of left ventricular function long term after arterial switch operation for transposition of the great arteries. Pediatr Cardiol. (2019) 40:188–93. doi: 10.1007/s00246-018-1977-6
114. Xie M, Zhang W, Cheng TO, Wang X, Lu X, Hu X. Left ventricular torsion abnormalities in patients after the arterial switch operation for transposition of the great arteries with intact ventricular septum. Int J Cardiol. (2013) 168:4631–7. doi: 10.1016/j.ijcard.2013.07.194
115. Shepard CW, Germanakis I, White MT, Powell AJ, Co-Vu J, Geva T. Cardiovascular magnetic resonance findings late after the arterial switch operation. Circ Cardiovasc Imaging. (2016) 9(9):e004618. doi: 10.1161/CIRCIMAGING.116.004618
116. Grotenhuis HB, Cifra B, Mertens LL, Riessenkampff E, Manlhiot C, Seed M, et al. Left ventricular remodelling in long-term survivors after the arterial switch operation for transposition of the great arteries. Eur Heart J Cardiovasc Imaging. (2019) 20:101–7. doi: 10.1093/ehjci/jey072
117. Suther KR, Hopp E, Geier O, Brun H, Nguyen B, Tomterstad AH, et al. Diffuse myocardial fibrosis in adolescents operated with arterial switch for transposition of the great arteries—a CMR study. Int J Cardiol. (2019) 276:100–6. doi: 10.1016/j.ijcard.2018.11.107
118. Wang C, Li VWY, So EKF, Cheung YF. Left ventricular stiffness in adolescents and young adults after arterial switch operation for complete transposition of the great arteries. Pediatr Cardiol. (2020) 41:747–54. doi: 10.1007/s00246-020-02305-2
119. Quing M, Görlach A, Schumacher K, Wötje M, Vazquez-Jimenez JF, Hess J, et al. The hypoxia-inducible factor HIF-1 promotes intramyocardial expression of VEGF in infants with congenital cardiac defects. Basic Res Cardiol. (2007) 102:224–32. doi: 10.1007/S00395-007-0639-2
120. Hölscher M, Schäfer K, Krull S, Farhat K, Hesse A, Silter M, et al. Unfavourable consequences of chronic cardiac HIF-1α stabilization. Cardiovasc Res. (2012) 94:77–86. doi: 10.1093/CVR/CVS014
121. Rathod RH, Prakash A, Powell AJ, Geva T. Myocardial fibrosis identified by cardiac magnetic resonance late gadolinium enhancement is associated with adverse ventricular mechanics and ventricular tachycardia late after fontan operation. J Am Coll Cardiol. (2010) 55:1721–8. doi: 10.1016/j.jacc.2009.12.036
122. Broberg CS, Prasad SK, Carr C, Babu-Narayan SV, Dimopoulos K, Gatzoulis MA. Myocardial fibrosis in Eisenmenger syndrome: a descriptive cohort study exploring associations of late gadolinium enhancement with clinical status and survival. J Cardiovasc Magn Reson. (2014) 16(1):32. doi: 10.1186/1532-429X-16-32
123. Kato A, Riesenkampff E, Yim D, Yoo SJ, Seed M, Grosse-Wortmann L. Pediatric Fontan patients are at risk for myocardial fibrotic remodeling and dysfunction. Int J Cardiol. (2017) 240:172–7. doi: 10.1016/j.ijcard.2017.04.073
124. Pisesky A, Reichert MJE, de Lange C, Seed M, Yoo SJ, Lam CZ, et al. Adverse fibrosis remodeling and aortopulmonary collateral flow are associated with poor Fontan outcomes. J Cardiovasc Magn Reson. (2021) 23(1):134. doi: 10.1186/s12968-021-00782-9
125. Kharabish A, Meierhofer C, Hadamitzky M, Nadjiri J, Martinoff S, Ewert P, et al. Long-standing cyanosis in congenital heart disease does not cause diffuse myocardial fibrosis. Pediatr Cardiol. (2018) 39:105–10. doi: 10.1007/s00246-017-1734-2
126. Buendía-Fuentes F, Gordon-Ramírez B, dos Subirà L, Merás P, Gallego P, González A, et al. Long-term outcomes of adults with single ventricle physiology not undergoing Fontan repair: a multicentre experience. Can J Cardiol. (2021) 38(7):1111–20. doi: 10.1016/J.CJCA.2021.06.001
127. Khairy P, Fernandes SM, Mayer JE, Triedman JK, Walsh EP, Lock JE, et al. Long-term survival, modes of death, and predictors of mortality in patients with Fontan surgery. Circulation. (2008) 117:85–92. doi: 10.1161/CIRCULATIONAHA.107.738559
128. Alsaied T, Moore RA, Lang SM, Truong V, Lubert AM, Veldtman GR, et al. Myocardial fibrosis, diastolic dysfunction and elevated liver stiffness in the Fontan circulation. Open Heart. (2020) 7. doi: 10.1136/openhrt-2020-001434
129. Clarke SA, Richardson WJ, Holmes JW. Modifying the mechanics of healing infarcts: is better the enemy of good? J Mol Cell Cardiol. (2016) 93:115–24. doi: 10.1016/J.YJMCC.2015.11.028
130. Lewis GA, Schelbert EB, Naish JH, Bedson E, Dodd S, Eccleson H, et al. Pirfenidone in heart failure with preserved ejection fraction-rationale and design of the PIROUETTE trial. Cardiovasc Drugs Ther. (2019) 33:461–70. doi: 10.1007/S10557-019-06876-Y
131. Brilla CG, Funck RC, Rupp H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation. (2000) 102:1388–93. doi: 10.1161/01.CIR.102.12.1388
132. López B, González A, Beaumont J, Querejeta R, Larman M, Díez J. Identification of a potential cardiac antifibrotic mechanism of torasemide in patients with chronic heart failure. J Am Coll Cardiol. (2007) 50:859–67. doi: 10.1016/J.JACC.2007.04.080
133. Puls M, Beuthner BE, Topci R, Vogelgesang A, Bleckmann A, Sitte M, et al. Impact of myocardial fibrosis on left ventricular remodelling, recovery, and outcome after transcatheter aortic valve implantation in different haemodynamic subtypes of severe aortic stenosis. Eur Heart J. (2020) 41:1903–14. doi: 10.1093/EURHEARTJ/EHAA033
134. Velu JF, Hirsch A, Matthijs Boekholdt S, Koch KT, Marije Vis M, Nils Planken R, et al. Myocardial fibrosis predicts adverse outcome after MitraClip implantation. Catheter Cardiovasc Interv. (2019) 93:1146–9. doi: 10.1002/CCD.27993
135. Treibel TA, Kozor R, Schofield R, Benedetti G, Fontana M, Bhuva AN, et al. Reverse myocardial remodeling following valve replacement in patients with aortic stenosis. J Am Coll Cardiol. (2018) 71:860–71. doi: 10.1016/J.JACC.2017.12.035
136. Liu B, Neil DAH, Bhabra M, Patel R, Barker TA, Nikolaidis N, et al. Reverse myocardial remodeling following valve repair in patients with chronic severe primary degenerative mitral regurgitation. JACC Cardiovasc Imaging. (2022) 15:224–36. doi: 10.1016/J.JCMG.2021.07.007
137. Boehm M, Tian X, Mao Y, Ichimura K, Dufva MJ, Ali K, et al. Delineating the molecular and histological events that govern right ventricular recovery using a novel mouse model of pulmonary artery de-banding. Cardiovasc Res. (2020) 116(10):1700–9. doi: 10.1093/CVR/CVZ310
138. Prisco SZ, Thenappan T, Prins KW. Treatment targets for right ventricular dysfunction in pulmonary arterial hypertension. JACC Basic Transl Sci. (2020) 5:1244. doi: 10.1016/J.JACBTS.2020.07.011
139. Andersen S, Birkmose Axelsen J, Ringgaard S, Randel Nyengaard J, Holm Nielsen S, Genovese F, et al. Pressure overload induced right ventricular remodeling is not attenuated by the anti-fibrotic agent pirfenidone. Pulm Circ. (2019) 9(2):1–13. doi: 10.1177/2045894019848659
140. Poble PB, Phan C, Quatremare T, Bordenave J, Thuillet R, Cumont A, et al. Therapeutic effect of pirfenidone in the sugen/hypoxia rat model of severe pulmonary hypertension. FASEB J. (2019) 33:3670–9. doi: 10.1096/FJ.201801659R
141. Boehm M, Arnold N, Braithwaite A, Pickworth J, Lu C, Novoyatleva T, et al. Eplerenone attenuates pathological pulmonary vascular rather than right ventricular remodeling in pulmonary arterial hypertension. BMC Pulm Med. (2018) 18:41. doi: 10.1186/S12890-018-0604-X
142. Bandula S, White SK, Flett AS, Lawrence D, Pugliese F, Ashworth MT, et al. Measurement of myocardial extracellular volume fraction by using equilibrium contrast-enhanced CT: validation against histologic findings. Radiology. (2013) 269:396–403. doi: 10.1148/RADIOLOGY.13130130
143. Nacif MS, Kawel N, Lee JJ, Chen X, Yao J, Zavodni A, et al. Interstitial myocardial fibrosis assessed as extracellular volume fraction with low-radiation-dose cardiac CT. Radiology. (2012) 264:876–83. doi: 10.1148/RADIOL.12112458
144. Tamarappoo B, Han D, Tyler J, Chakravarty T, Otaki Y, Miller R, et al. Prognostic value of computed tomography-derived extracellular volume in TAVR patients with low-flow low-gradient aortic stenosis. JACC Cardiovasc Imaging. (2020) 13:2591–601. doi: 10.1016/J.JCMG.2020.07.045
145. Scully PR, Patel KP, Klotz E, Augusto JB, Thornton GD, Saberwal B, et al. Myocardial fibrosis quantified by cardiac CT predicts outcome in severe aortic stenosis after transcatheter intervention. JACC Cardiovasc Imaging. (2022) 15:542. doi: 10.1016/J.JCMG.2021.10.016
Keywords: myocardial fibrosis, fibrosis, congenital heart disease, cardiac magnetic resonance (CMR), collagen biomarkers, tetralogy of Fallot, systemic right ventricle, myocardial interstitial fibrosis
Citation: Gordon B, González-Fernández V and Dos-Subirà L (2022) Myocardial fibrosis in congenital heart disease. Front. Pediatr. 10:965204. doi: 10.3389/fped.2022.965204
Received: 9 June 2022; Accepted: 18 October 2022;
Published: 18 November 2022.
Edited by:
Gerhard-Paul Diller, University Hospital Münster, GermanyReviewed by:
Craig Broberg, Oregon Health and Science University, United States© 2022 Gordon, González-Fernández and Dos-Subirà. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Víctor González-Fernández dmljdG9yLmdvbmZlckBnbWFpbC5jb20=
Specialty Section: This article was submitted to Pediatric Cardiology, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.