
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 14 November 2022
Sec. Pediatric Cardiology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.947963
 Junmin Zheng1,†
Junmin Zheng1,† Zhuangzhuang Huang1,†Shan Hou1Xunwei Jiang1Yongwei Zhang1Wei Liu1
Zhuangzhuang Huang1,†Shan Hou1Xunwei Jiang1Yongwei Zhang1Wei Liu1 Jia Jia2Yun Li1Xiaomin Sun1Lijian Xie1*Xiaopei Zhao1*Cuilan Hou1*
Jia Jia2Yun Li1Xiaomin Sun1Lijian Xie1*Xiaopei Zhao1*Cuilan Hou1* Tingting Xiao1*
Tingting Xiao1*
Hypertrophic cardiomyopathy (HCM) is an autosomal dominant cardiomyopathy, which is one of the most common reasons for cardiac arrest in children or adolescents. It is characterized by ventricular hypertrophy (usually left ventricle), small ventricular cavity, and reduced ventricular diastolic compliance found by echocardiography in the absence of abnormal load (such as hypertension or aortic stenosis). HCM is usually caused by mutations in genes encoding sarcomere or sarcomere-related genes. Whole exome sequencing (WES) is performed to identify probable causative genes. Through WES, we identified LIM domain-binding protein 3 (LDB3) mutations (R547Q and P323S) respectively in an 11-year-old HCM girl and a 6-year-old HCM boy. Neural network analyses showed that the LDB3 (R547Q and P323S) mutation decreased its protein stability, with confidence scores of −0.9211 and −0.8967. The STRUM server also confirmed that the mutation decreased its protein stability. Thus, LDB3 mutation may be associated with heritable HCM. To our knowledge, this is the first time to report LDB3 heterozygous variants (R547Q and P323S) responsible for heritable HCM.
Hypertrophic cardiomyopathy (HCM) is an autosomal dominant primary myocardial disease, the characteristics of which are ventricular hypertrophy (usually left ventricle), small ventricular cavity, and reduced ventricular diastolic compliance found by echocardiography in the absence of abnormal load (such as hypertension or aortic stenosis) (1). Myocyte disorder, extracellular matrix changes, microvascular remodeling, and interstitial fibrosis are the main histological features of HCM (2, 3). The incidence rate of HCM is about 1/500–1/200, and it can affect people of any age. Many patients may have no obvious clinical manifestations such as signs or symptoms and cannot be diagnosed early. Therefore, it may be an important cause of cardiac arrest in children or adolescents (1, 4). Of course, one of the primary approaches to diagnosing HCM is transthoracic echocardiography. In addition to ventricular wall hypertrophy, it can also be combined with a series of complex manifestations, such as mitral valve abnormality and left ventricular outflow tract obstruction (5). HCM is usually caused by mutations in genes encoding sarcomere or sarcomere-related genes. More than 1,400 disease-causing genes in HCM have been identified (6). MYBPC3 and MYH7 are generally the predominant HCM disease-causing genes, accounting for 50%–70%, followed by TNNT2, TNNI3, MYL2, MYL3, TPM1, and ACTC1 (6). In addition, genes encoding Z-disk components and Ca2+ homeostasis factors, such as TCAP, CSRP3, ACTN2, and JPH2, can also cause HCM (6). It is well reported that most cases of HCM are caused by single-nucleotide polymorphism in genes encoding cardiac proteins (7, 8), but the mutation-related mechanism leading to HCM is not clearly understood. However, it has been reported that genetic diversity may lead to multiple gene mutations, and these genes may jointly affect the patient phenotype (9). Molecular genetic research studies provide important insights into HCM pathogenesis and support a new perspective for the diagnosis and treatment of the disease (5).
In 2014, LDB3 gene was first reported to be closely related to HCM (10). Fratev et al. first reported a new ZASP PDZ mutation (Gly54Ser) in HCM (10). They also demonstrated that the mutation significantly affects the interactions between ZASP PDZ domains and their binding partners (10). It is suggested that ZASPG54S may be related to HCM, and LDB3 gene should be considered in general HCM gene detection (10).
In this study, we collected HCM pedigrees with two affected and five unaffected members. Whole exome sequencing (WES) was utilized to identify possible disease-causing genes or variants. Paired end reading was aligned with the GRCh37/hg19 human reference sequence. Through comprehensive ClinVar software and GATK analyses, BAM and VCF files were produced. LDB3 gene heterozygous mutation R547Q in an 11-year-old girl and P323S in a 6-year-old boy with HCM were identified. Related families were also employed to WES in order to confirm possible disease-causing genes or variants. Our results indicate that LDB3 mutation may be associated with HCM and should be screened in prospective clinical practice to encourage early intervention.
An 11-year-old girl was hospitalized with palpitation after exercise for 6 years. There was no special sign in her physical examination. Her 12-lead electrocardiogram (ECG) in resting time showed sinus rhythm and ventricular preexcitation, and her echocardiogram showed slight left ventricular hypertrophy after treatment (Figure 1A), although her left atrium and left ventricle were dilated, and her left ventricular ejection fraction was 25% via echocardiogram (Figure 1B) at the first time. The patient was given myocardial nutrition, cardiotonic diuresis, and her condition improved. Another 6-year-old boy was admitted with left ventricular hypertrophy. There was no fever, chest pain, chest tightness, palpitation, syncope, or other discomforts. There was also no special sign on his physical examination. His 12-lead ECG in resting time was normal, and echocardiogram revealed slight left ventricular hypertrophy (Figures 1C,D). He was treated with myocardial nutrition, and his condition improved. We also checked the biochemical metabolism, myocardial enzyme, and cardiac computed tomography angiography (CTA), and both were normal. The two patients should rest to avoid infection and fatigue.
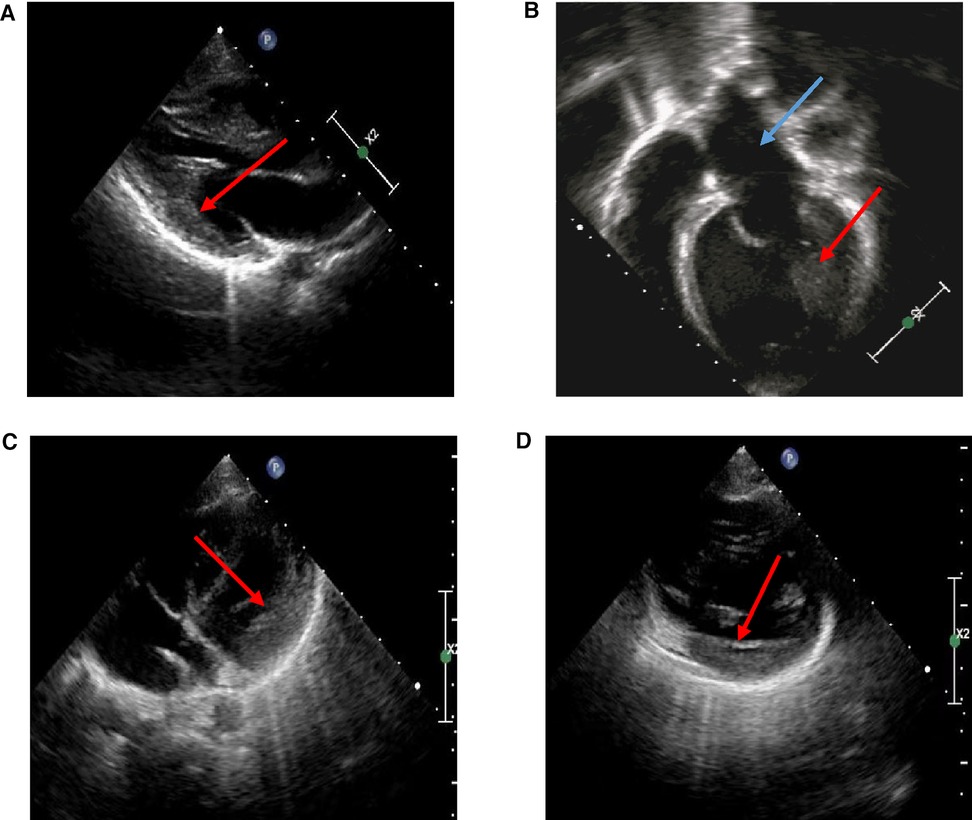
Figure 1. Echocardiography in HCM probands. (A) Echocardiography of a proband: slight left ventricular hypertrophy at later observation (red arrow). (B) Echocardiography of a proband: enlargement of left atrial (blue arrow) and left ventricle (red arrow) at first time. (C,D) Echocardiography of a proband: slight left ventricular hypertrophy (red arrow).
The construction of DNA libraries and WES were performed using the manufacturer's instructions. Genomic DNA was extracted from peripheral venous blood samples of children and their families via a whole-blood genomic DNA Extraction Kit (Tiangen, China). According to the manufacturer's instructions, 1 µg DNA in WES was prepared by using TruSeq DNA LT Sample Prep kit v2, hybridized with Nimblegen SeqCap EZ Exome v3 (Roche) and then matched and sequenced (2 × 100 bp) with TruSeq v3 chemistry on Illumina HiSeq 2500.
LDB3 mutation was confirmed by Sanger sequencing. Briefly, primers were designed that cover the mutation sequence. LDB3 mutation (R547Q) forward: 5′-CCCAACTATAACCCTGCACCCTC-3′and reverse: 5′-GCCCTGTAAACCTACTGACAAGC-3′. LDB3 mutation (P323S) forward: 5′-GCTCCCTTGACCTGTTGTCT-3′ and reverse: 5′-GGCCCTAACTACCTTGGACACA-3′. PCR products were analyzed on agarose gels and purified with the QIAqick kit (Qiagen, United States). Sanger sequencing is supported by ABI3500 (Applied Biosystems, United States) platform of Suzhou Hong Xun Biotechnology Co., Ltd.
Specifically, self-developed software was used to filter the sequencing data. Then, the filtered data were compared with a human genome database (GRCh 37/Hg 19) via BWA-0.710 software. Base mutations and insertion–deletion mutations in sequencing were screened and identified, compared with 370 whole exome sequencing samples in our hospital as well as 1000 Genomes Project, gnomAD, HGMD, Exome Variant Server, Exome Consortium databases, ClinVar (http:// www.ncbi.nlm.nih.gov/clinvar), and OMIM. Mutation sites were evaluated using COBALT homology alignment for evaluating amino acid sequences from different species. GERP (genomic evolutionary rate profiling), Mutation Taster (www.mutationtaster.org), combined annotation-dependent depletion (CADD), SIFT (Sorting Intolerant from Tolerant; http://siftdna.org/), and Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/) were used to predict mutations among conservative species, the influence of protein structure and function. Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org) was used to test the frequency of variations. Polymerase chain reaction was used for amplification of rare mutations. Frequency between affected family members and healthy individuals and the presence or absence of variants were screened with Illumina Variant Studio. Experimental methods and data analysis were referred to the previous study (11).
Real-time-PCR (RT-PCR) was utilized to detect LDB3 mRNA levels. Whole-blood samples (200 µl) were used to extract RNA according to the protocol (PrimeScript™ RT Master Mix, Takara). Primers were designed to explore whether the mutation could alter its expression. Related primers are shown in Table 1.
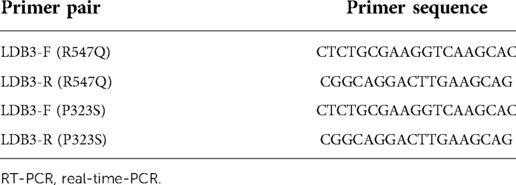
Table 1. Primers used for vector construction and RT-PCR.
As Figures 2A,B showed, 80,192 variants were detected and further annotated and filtered by Ingenuity Variant Analysis. According to Exome Aggregation Consortium, 1000 Genomes Project, Exome Sequencing Project, or gnomAD, 78,497 common variants were filtered and eliminated from their frequencies (minor allele frequency (MAF) < 0.01). Four variants in four genes (TGFB3, LMNA, MYH7, and LDB3) were determined and identified through a thorough analysis. Finally, LDB3 mutation was detected and selected after rigorous analysis, which was linked to the HCM phenotypes (Figure 2B). The same filtration process was undertaken with the other pedigree, and LDB3 mutation was also linked to the HCM phenotype (Figures 2C,D). These rare phenotype-related variants were classified with the American College of Medical Genetics and Genomics/Association for Molecular Pathology guidelines.
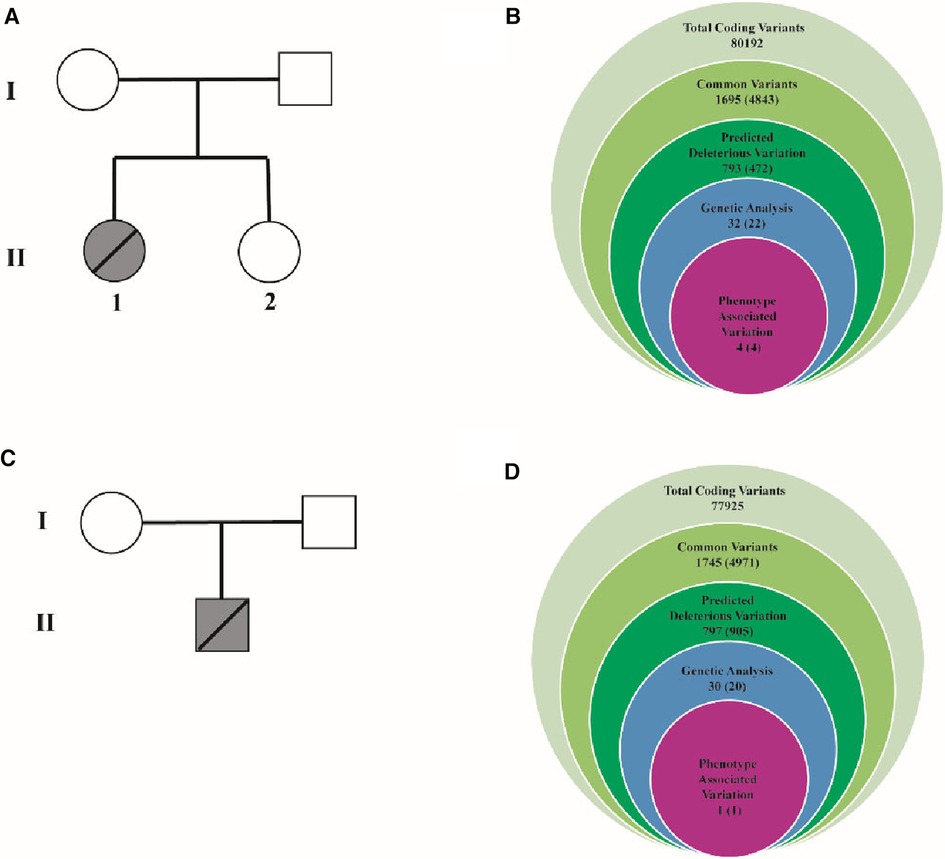
Figure 2. Family pedigrees and filtering processes for WES data. (A) One pedigree consists of one proband and three members. I-1 represents the proband's mother. I-2 represents the proband's father. II-1 represents the proband. II-2 represents the proband's sister. (B) The filtering process for WES data. It contains 80,192 total coding variants. Then, 1,695 common variants, 793 deleterious variations, 32 genetic analyses, and final 4 associated with this phenotype variation were filtered. TGFB3:NM_001329938:exon2:c.C487T: p.R163W, LMNA:NM_005572:exon10:c.G1712A: p.R571H, MYH7:NM_000257: exon3:c.C77T: p.A26V were deleted. (C) One pedigree consists of one proband. I-1 represents the proband's mother. I-2 represents the proband's father. II-1 represents the proband. (D) The filtering process for WES data. It contains 77,925 total coding variants. Then, 1, 745 common variants, 797 deleterious variations, 30 genetic analyses, final 1 associated with this phenotype variation were filtered.
Heterozygous mutations of LDB3 gene (R547Q and P323S) were identified in these two HCM pedigrees via WES. The mutation site c.1655G > A (p.R547Q) was located in the 12th exome of LDB3. The mutation site c.967C > T (p.P323S) was located in the 10th exome of LDB3 (Figure 3A). The proband and her father were confirmed to be heterozygous carriers of 1,655 G > A hybridization. Her mother and sister were confirmed to be homozygous negative of the mutation (Figure 3B). The other proband was confirmed to be heterozygous carrier of 967 C > T hybridization. His parents were homozygous negative of the mutation as confirmed by Sanger sequencing (Figure 3C).
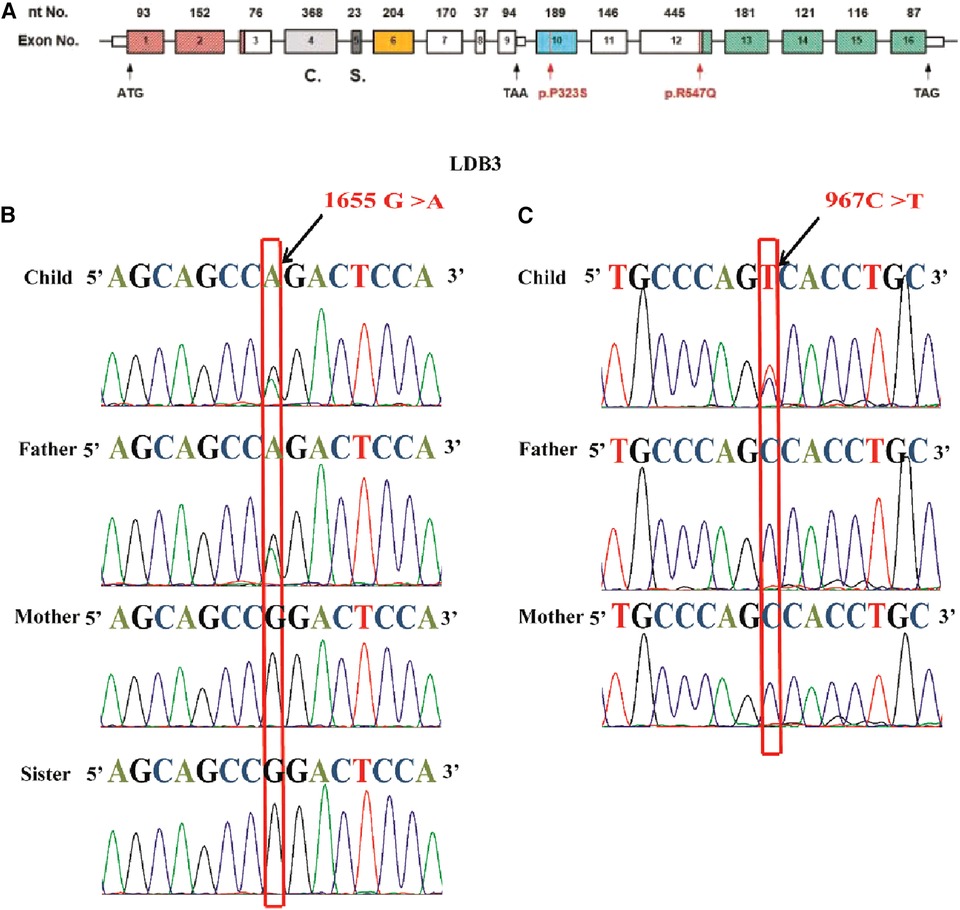
Figure 3. The LDB3 mutation site and Sanger sequencing. (A) Human LDB3 gene maps to chromosome 10q23.2 and contains 16 exomes. The base pair mutation site is c.967C > T (p.P323S), which is located in the 10th exome of Mybcp3. The base pair mutation site is c.1655G > A, which is located in the 12th exome of Mybcp3. (B) The proband and her father were confirmed to be heterozygous carriers of 1655 G > A hybridization. Her mother and sister were homozygous negative of the mutation. (C) The other proband was confirmed to be heterozygous carriers of 967 C > T hybridization. His parents were homozygous negative of the mutation as showed through Sanger sequencing.
LDB3 mutants were analyzed to further explore the possibility that the function of LDB3 mutants (R547Q and P323S) affected the occurrence of diseases (Figures 4A,B). LDB3 heterozygous variant (P323S) was located in a domain of unknown function, and LDB3 heterozygous variant (R547Q) was in the LIM zinc-binding domain. Importantly, this mutation (c.1655 G > A) was located in the LDB3 conserved region (Figure 4C), indicating that the mutation may affect its protein function. Neural network analyses showed that the LDB3 (R547Q and P323S) mutation decreased its protein stability, with confidence scores of −0.9211 and −0.8967. The STRUM server also confirmed that the mutation affected LDB3 protein stability (Figures 4D,E). In addition, protein stability was confirmed by a number of web-based prediction tools such as I-Mutant and MuPRO, which also showed that the mutation affected LDB3 protein stability (Table 2). The above data indicate that LDB3 heterozygous variants (R547Q and P323S) may disrupt its protein structure and ability.
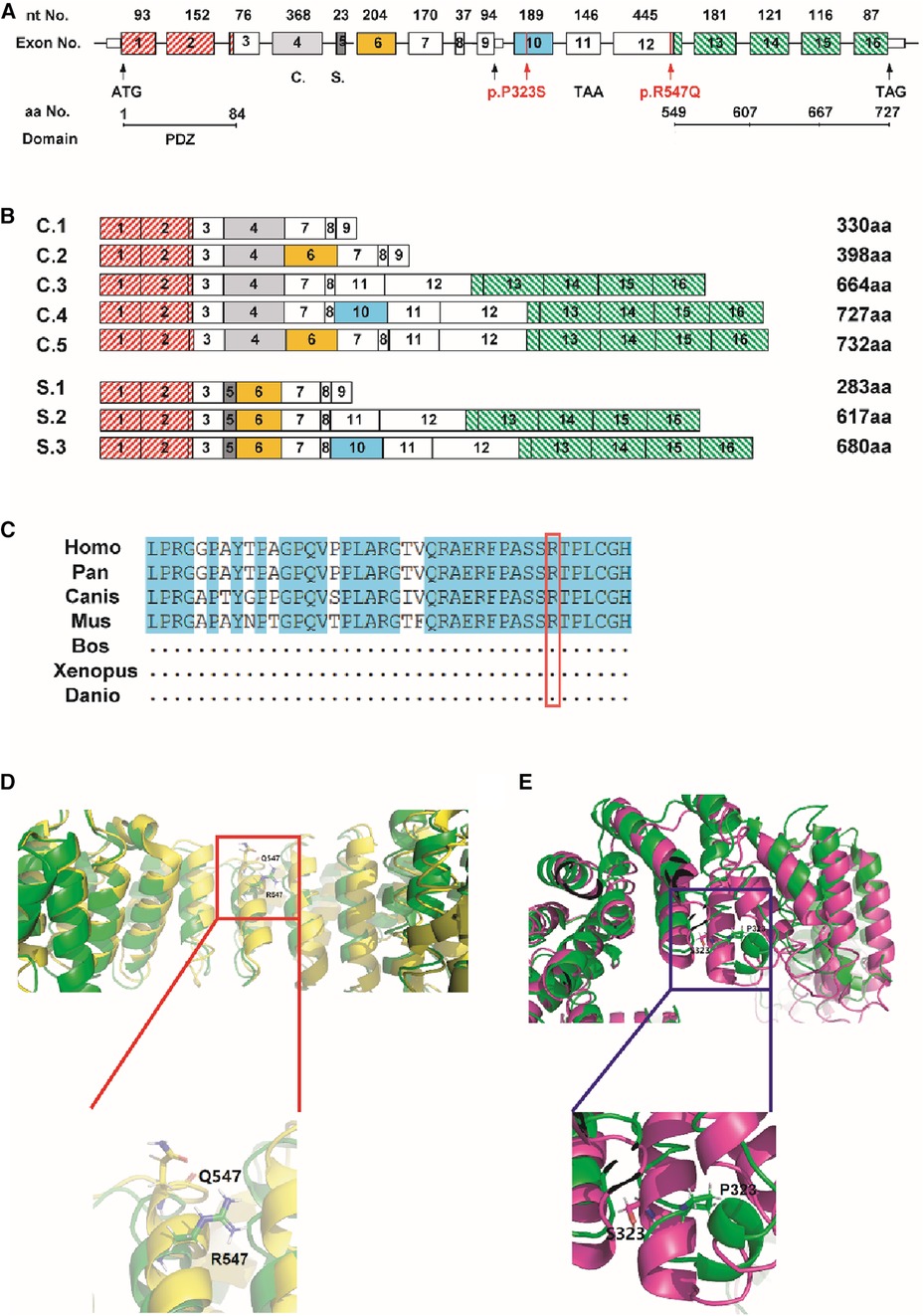
Figure 4. Prediction of the mutation on its functional. (A,B) The location of the mutation (c.967C > T, c.1655G > A) in the LDB3 gene. (C) c.967C > T mutation region locates in a highly conserved area of cross-species. (D,E) STRUM server indicated that the P323S and R547Q mutation may affect LDB3 protein stability.
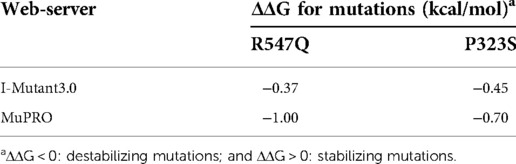
Table 2. Predictions using web-based tools.
In this study, we collected and analyzed the clinical phenotype and genotype of two mutated HCM pedigrees to investigate the potential disease-causing variant. Two interesting findings are as follows: (1) R547Q and P323S heterozygous mutations of LDB3 are first reported in HCM pedigrees; (2) Protein modeling assay indicates that these two LDB3 mutations (R547Q and P323S) may reduce its protein stability, which may play a crucial role for heritable HCM.
As a genetic myocardial disease, the pathogenic mechanism of HCM has not been fully understood yet. Up to now, several pathogenic hypotheses of HCM have been reported. The first theory suggests that mutations in genes encoding sarcomere proteins (such as β-MHC) and intracellular cytosomal proteins (such as titin) lead to a decrease in the rate and contraction velocity of muscle cells, alter the activity and mechanical effects of neurohumors, leading to compensatory hypertrophy of the myocardium (5). “Energy compromise” is considered a possible stimulant for the development of cardiac hypertrophy in HCM (12). Briefly, it proposed that major mutated proteins (β-MHC, cTnT, and MyBPC) cause energy depletion at certain key sites of muscle cells, a relative decrease in PCr/ATP, and are independent of cardiac hypertrophy. The third theory proposes that the mutation results in an invalid allele with haplotype dysfunction. That is, the production of normal sarcomere protein is reduced, and the composition of thick and thin sarcomere is out of balance, leading to changes in sarcomere structure and function (13, 14).
Genetic screening is an ideal tool for identification nucleotide changes that cause HCM. Although HCM has been attributed to monogenic interference in sarcomeric genes such as MYBPC3 and MYH7, significant phenotypic variation is often present even in a single family (15). This variability may be attributed to gene differential expression, protein–protein interaction, and protein modification variation (15). Recently, more than 1,400 HCM-related mutations have been identified, and they are almost sarcomeric proteins and Z-disk proteins (10). Fratev et al. first reported a new ZASP PDZ mutation (Gly54Ser) in HCM, and they demonstrated that this mutation significantly affects the interactions between ZASP PDZ domains and their binding partners (10). Our results also confirmed the occurrence of LDB3 gene mutation in HCM, and the heterozygous mutation site (R547Q and P323S) has not been reported previously. An RT-PCR assay was utilized to analyze LDB3 expression among the proband and her or his families. The LDB3 mutation site (R547Q and P323S) did not alter its mRNA expression in the two pedigrees (Tables 3, 4).

Table 3. Relative expression levels of LDB3 mRNA in one HCM family pedigree [LDB3 (R547Q)].

Table 4. Relative expression levels of LDB3 mRNA in one HCM family pedigree [LDB3 (P323S)].
ZASP is a sarcomeric protein encoded by LDB3 and specifically expressed in human heart and skeletal muscle (16). It is well known that ZASP variants are associated with myopathies such as dilated cardiomyopathy (DCM) (17), left ventricular non-compaction cardiomyopathy (LVNC) (18, 19), and HCM (20). LDB3 gene belongs to the PDZ-LIM domain protein family; its N-terminal contains a PDZ domain and C-terminal contains three LIM domains (21, 22). It has been reported that the PDZ domain interacts with the C terminus of α-actinin-2, and the LIM domains bind to protein kinase C and potentially have a role in protein kinase C-mediated signaling pathways (22, 23). As a sarcomeric protein, LDB3 interacts with Z-line proteins α-actinin-2, myotilin, and signaling molecules such as protein kinases (24), which indicates that LDB3 might play a crucial role in cardiomyopathy.
To further explore the impact of LDB3 variants (R547Q and P323S) on the occurrence of the HCM disease, we mapped the variant to the LDB3 protein structure. It is reported that variants locating within specific functional domains or protein translation modification sites can alter the protein function such as protein–ligand binding or protein–protein interaction (8, 25). In this study, LDB3 variants (R547Q and P323S) locate in the domain of unknown function and LIM zinc-binding domain, and the two heterozygosity mutations were both yet reported previously. It was reported that α-actinin-2 interacted with both PDZ and LIM domains (23, 26). Protein modeling assay indicates that the LDB3 mutation reduced its protein hydrophobicity and stability. Thus, the LDB3 mutation may disrupt its interaction with α-actinin-2, which plays a critical role in the heart muscle regulation. Previous research pointed that the actin-binding site in ZASP is also expressed in the heart muscle (27). Up to 20% of cardiac muscle actin consists of the skeletal muscle α-actin isoform (28). Multiple ZASP isoforms may play unique roles in maintaining the Z-disc structure by interacting with the actin in heart as well as skeletal muscle (29). The actin-binding domain is between the modular protein interacting PDZ and LIM domains. It is worth that LDB3 mutation (P323S) was located between PDZ and LIM domains. Therefore, LDB3 gene mutation can affect the binding of this domain to actin and affect the normal contractile or diastolic function of the cardiac or skeletal muscle.
Genetic pathogenic mutation and consideration of family history are necessary components of HCM diagnostic criteria. In this study, we identified two heterozygous mutation sites of LDB3 that were associated with HCM. Two pedigrees were confirmed with LDB3 heterozygous mutation (R547Q and P323S), indicating that LDB3 mutation may be a novel disease-causing variant for heritable HCM. Systematic analysis not only improves our understanding of this disease etiology but also contributes to clinical and prenatal diagnosis.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
TX and CH designed the project. JZ, ZH, LX, and XZ provided the bioinformatics analysis and wrote the manuscript with input from SH, XJ, YZ, WL, JJ, YL, and XS. All authors read and approved the final manuscript.
This work is supported by Shanghai Jiao Tong University medical technology crossing project (YG2021ZD26), National Natural Science Foundation of China (NSFC) (no. 81900437, 82170518), and Shanghai Children's hospital (2019YQ06, 2020YGZQ10), Talent Program of Shanghai Children's Hospital, and Shanghai Collaborative Innovation Center for Translational Medicine (TM201922).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
LDB3, LIM domain-binding protein 3; HCM, hypertrophic cardiomyopathy; WES, whole exome sequencing; ECG, electrocardiogram; CTA, cardiac computed tomography angiography; RT-PCR, real-time-PCR; DCM, dilated cardiomyopathy; LVNC, left ventricular non-compaction cardiomyopathy
1. Ommen SR, Semsarian C. Hypertrophic cardiomyopathy: a practical approach to guideline directed management. Lancet. (2021) 398(10316):2102–8. doi: 10.1016/S0140-6736(21)01205-8
2. Muresan ID, Agoston-Coldea L. Phenotypes of hypertrophic cardiomyopathy: genetics, clinics, and modular imaging. Heart Fail Rev. (2021) 26(5):1023–36. doi: 10.1007/s10741-020-09931-1
3. Zampieri M, Argiro A, Marchi A, Berteotti M, Targetti M, Fornaro A, et al. Mavacamten, a novel therapeutic strategy for obstructive hypertrophic cardiomyopathy. Curr Cardiol Rep. (2021) 23(7):79. doi: 10.1007/s11886-021-01508-0
4. Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, et al. Management of hypertrophic cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. (2022) 79(4):390–414. doi: 10.1016/j.jacc.2021.11.021
5. Fatkin D, Graham RM. Molecular mechanisms of inherited cardiomyopathies. Physiol Rev. (2002) 82(4):945–80. doi: 10.1152/physrev.00012.2002
6. Tran Vu MT, Nguyen TV, Huynh NV, Nguyen Thai HT, Pham Nguyen V, Ho Huynh TD. Presence of hypertrophic cardiomyopathy related gene mutations and clinical manifestations in Vietnamese patients with hypertrophic cardiomyopathy. Circ J. (2019) 83(9):1908–16. doi: 10.1253/circj.CJ-19-0190
7. Popa-Fotea NM, Micheu MM, Bataila V, Scafa-Udriste A, Dorobantu L, Scarlatescu AI, et al. Exploring the continuum of hypertrophic cardiomyopathy-from DNA to clinical expression. Medicina (Kaunas). (2019) 55(6):299. doi: 10.3390/medicina55060299
8. Pradeep R, Akram A, Proute MC, Kothur NR, Georgiou P, Serhiyenia T, et al. Understanding the genetic and molecular basis of familial hypertrophic cardiomyopathy and the current trends in gene therapy for its management. Cureus. (2021) 13(8):e17548. doi: 10.7759/cureus.17548
9. Li L, Bainbridge MN, Tan Y, Willerson JT, Marian AJ. A potential oligogenic etiology of hypertrophic cardiomyopathy: a classic single-gene disorder. Circ Res. (2017) 120(7):1084–90. doi: 10.1161/CIRCRESAHA.116.310559
10. Fratev F, Mihaylova E, Pajeva I. Combination of genetic screening and molecular dynamics as a useful tool for identification of disease-related mutations: ZASP PDZ domain G54S mutation case. J Chem Inf Model. (2014) 54(5):1524–36. doi: 10.1021/ci5001136
11. Xie L, Hou C, Jiang X, Zhao J, Li Y, Xiao T. A compound heterozygosity of Tecrl gene confirmed in a catecholaminergic polymorphic ventricular tachycardia family. Eur J Med Genet. (2019) 62(7):103631. doi: 10.1016/j.ejmg.2019.01.018
12. Crilley JG, Boehm EA, Blair E, Rajagopalan B, Blamire AM, Styles P, et al. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J Am Coll Cardiol. (2003) 41(10):1776–82. doi: 10.1016/S0735-1097(02)03009-7
13. Zolk O, Schenke C, Sarikas A. The ubiquitin-proteasome system: focus on the heart. Cardiovasc Res. (2006) 70(3):410–21. doi: 10.1016/j.cardiores.2005.12.021
14. Brown DI, Parry TL, Willis MS. Ubiquitin ligases and posttranslational regulation of energy in the heart: the hand that feeds. Compr Physiol. (2017) 7(3):841–62. doi: 10.1002/cphy.c160024
15. Gao J, Collyer J, Wang M, Sun F, Xu F. Genetic dissection of hypertrophic cardiomyopathy with myocardial RNA-seq. Int J Mol Sci. (2020) 21(9):3040 doi: 10.3390/ijms21093040.
16. Lv J, Pan Z, Chen J, Xu R, Wang D, Huang J, et al. Phosphoproteomic analysis reveals downstream PKA effectors of AKAP cypher/ZASP in the pathogenesis of dilated cardiomyopathy. Front Cardiovasc Med. (2021) 8:753072. doi: 10.3389/fcvm.2021.753072
17. Gerull B, Gramlich M, Atherton J, McNabb M, Trombitas K, Sasse-Klaassen S, et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet. (2002) 30(2):201–4. doi: 10.1038/ng815
18. Xing Y, Ichida F, Matsuoka T, Isobe T, Ikemoto Y, Higaki T, et al. Genetic analysis in patients with left ventricular noncompaction and evidence for genetic heterogeneity. Mol Genet Metab. (2006) 88(1):71–7. doi: 10.1016/j.ymgme.2005.11.009
19. Moric-Janiszewska E, Markiewicz-Loskot G. Genetic heterogeneity of left-ventricular noncompaction cardiomyopathy. Clin Cardiol. (2008) 31(5):201–4. doi: 10.1002/clc.20202
20. Theis JL, Bos JM, Bartleson VB, Will ML, Binder J, Vatta M, et al. Echocardiographic-determined septal morphology in Z-disc hypertrophic cardiomyopathy. Biochem Biophys Res Commun. (2006) 351(4):896–902. doi: 10.1016/j.bbrc.2006.10.119
21. Zhou Q, Ruiz-Lozano P, Martone ME, Chen J. Cypher, a striated muscle-restricted PDZ and LIM domain-containing protein, binds to alpha-actinin-2 and protein kinase C. J Biol Chem. (1999) 274(28):19807–13. doi: 10.1074/jbc.274.28.19807
22. Cassandrini D, Merlini L, Pilla F, Cenni V, Santi S, Faldini C, et al. Protein aggregates and autophagy involvement in a family with a mutation in Z-band alternatively spliced PDZ-motif protein. Neuromuscul Disord. (2021) 31(1):44–51. doi: 10.1016/j.nmd.2020.11.008
23. Faulkner G, Pallavicini A, Formentin E, Comelli A, Ievolella C, Trevisan S, et al. ZASP: a new Z-band alternatively spliced PDZ-motif protein. J Cell Biol. (1999) 146(2):465–75. doi: 10.1083/jcb.146.2.465
24. Zheng M, Cheng H, Li X, Zhang J, Cui L, Ouyang K, et al. Cardiac-specific ablation of Cypher leads to a severe form of dilated cardiomyopathy with premature death. Hum Mol Genet. (2009) 18(4):701–13. doi: 10.1093/hmg/ddn400
25. Marian AJ. Molecular genetic basis of hypertrophic cardiomyopathy. Circ Res. (2021) 128(10):1533–53. doi: 10.1161/CIRCRESAHA.121.318346
26. Au Y, Atkinson RA, Guerrini R, Kelly G, Joseph C, Martin SR, et al. Solution structure of ZASP PDZ domain; implications for sarcomere ultrastructure and enigma family redundancy. Structure. (2004) 12(4):611–22. doi: 10.1016/j.str.2004.02.019
27. Vatta M, Mohapatra B, Jimenez S, Sanchez X, Faulkner G, Perles Z, et al. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol. (2003) 42(11):2014–27. doi: 10.1016/j.jacc.2003.10.021
28. Vandekerckhove J, Bugaisky G, Buckingham M. Simultaneous expression of skeletal muscle and heart actin proteins in various striated muscle tissues and cells. A quantitative determination of the two actin isoforms. J Biol Chem. (1986) 261(4):1838–43. doi: 10.1016/S0021-9258(17)36017-9
29. Lin X, Ruiz J, Bajraktari I, Ohman R, Banerjee S, Gribble K, et al. Z-disc-associated, alternatively spliced, PDZ motif-containing protein (ZASP) mutations in the actin-binding domain cause disruption of skeletal muscle actin filaments in myofibrillar myopathy. J Biol Chem. (2014) 289(19):13615–26. doi: 10.1074/jbc.M114.550418
Keywords: hypertrophic cardiomyopathy, LIM domain-binding protein 3, whole exome sequencing, gene mutation, protein stability
Citation: Zheng J, Huang Z, Hou S, Jiang X, Zhang Y, Liu W, Jia J, Li Y, Sun X, Xie L, Zhao X, Hou C and Xiao T (2022) Case Report: Novel LIM domain-binding protein 3 (LDB3) mutations associated with hypertrophic cardiomyopathy family. Front. Pediatr. 10:947963. doi: 10.3389/fped.2022.947963
Received: 19 May 2022; Accepted: 28 September 2022;
Published: 14 November 2022.
Edited by:
Sameh M. Said, Westchester Medical Center, United StatesReviewed by:
Liqun Sun, University of Toronto, Canada© 2022 Zheng, Huang, Hou, Jiang, Zhang, Liu, Jia, Li, Sun, Xie, Zhao, Hou and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lijian Xie bmFpamlsZWl4QGFsaXl1bi5jb20= Xiaopei Zhao emhhb3hwZG9jdG9yQDEyNi5jb20= Cuilan Hou aG91Y2w4OEBzaW5hLmNu Tingting Xiao dHR4aWFvMjAxN0AxNjMuY29t
†These authors share first authorship
Specialty Section: This article was submitted to Pediatric Cardiology, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.