 Rodrigo Tzovenos Starosta1*
Rodrigo Tzovenos Starosta1* Ying-Chen Claire Hou2Katelyn Leestma1Prapti Singh1
Ying-Chen Claire Hou2Katelyn Leestma1Prapti Singh1 Luke Viehl3Linda Manwaring1Jorge Luis Granadillo1Molly C. Schroeder2Jamie N. Colombo4Halana Whitehead3Patricia Irene Dickson1
Luke Viehl3Linda Manwaring1Jorge Luis Granadillo1Molly C. Schroeder2Jamie N. Colombo4Halana Whitehead3Patricia Irene Dickson1 Monica L. Hulbert5
Monica L. Hulbert5 Hoanh Thi Nguyen1
Hoanh Thi Nguyen1- 1Division of Clinical Genetics and Genomics, Department of Pediatrics, Washington University School of Medicine, St. Louis, MO, United States
- 2Department of Pathology and Immunology, Washington University School of Medicine, St. Louis, MO, United States
- 3Division of Newborn Medicine, Department of Pediatrics, Washington University School of Medicine, St. Louis, MO, United States
- 4Division of Pediatric Cardiology, Department of Pediatrics, Washington University School of Medicine, St. Louis, MO, United States
- 5Division of Pediatric Hematology and Oncology, Department of Pediatrics, Washington University School of Medicine, St. Louis, MO, United States
Infantile-onset Pompe disease (IOPD) is a rare, severe disorder of lysosomal storage of glycogen that leads to progressive cardiac and skeletal myopathy. IOPD is a fatal disease in childhood unless treated with enzyme replacement therapy (ERT) from an early age. Sickle cell anemia (SCA) is a relatively common hemoglobinopathy caused by a specific variant in the hemoglobin beta-chain. Here we report a case of a male newborn of African ancestry diagnosed and treated for IOPD and SCA. Molecular testing confirmed two GAA variants, NM_000152.5: c.842G>C, p.(Arg281Pro) and NM_000152.5: c.2560C>T, p.(Arg854*) in trans, and homozygosity for the HBB variant causative of SCA, consistent with his diagnosis. An acute neonatal presentation of hypotonia and cardiomyopathy required ERT with alglucosidase alfa infusions preceded by immune tolerance induction (ITI), as well as chronic red blood cell transfusions and penicillin V potassium prophylaxis for treatment of IOPD and SCA. Clinical course was further complicated by multiple respiratory infections. We review the current guidelines and interventions taken to optimize his care and the pitfalls of those guidelines when treating patients with concomitant conditions. To the best of our knowledge, no other case reports of the concomitance of these two disorders was found. This report emphasizes the importance of newborn screening, early intervention, and treatment considerations for this complex patient presentation of IOPD and SCA.
Introduction
Pompe disease (MIM #232300), also known as “glycogen storage disease type II”, is an autosomal recessive inborn error of glycogen metabolism that leads to lysosomal storage of undigested glycogen in muscle tissues. It is caused by a deficiency of acid alpha-glucosidase (GAA; EC 3.2.1.20), coded by GAA on chromosome 17q25.3. Pompe disease is generally categorized according to the age of onset of clinical manifestations: infantile-onset Pompe disease (IOPD) is characterized by onset of generalized hypotonia, macroglossia, and left ventricular hypertrophy before 12 months of age, and it generally leads to death by respiratory and cardiac failure before the second year of age unless early enzyme replacement therapy (ERT) is instituted (1, 2). Late-onset Pompe disease (LOPD), on the other hand, presents as proximal skeletal muscle weakness that progresses to involve bulbar, respiratory, and distal skeletal musculature as well as visceral smooth muscle, with relative sparing of cardiac muscle (3). Alglucosidase alfa, a recombinant form of acid alpha-glucosidase, is the only ERT modality currently available for the treatment of IOPD and has been shown to increase overall survival and ventilator-free survival, as well as to decrease left ventricular mass and increase left ventricular ejection fraction in patients with classic IOPD (4). The standard, FDA-approved alglucosidase alfa dose is 20 mg/kg every 2 weeks (4), although there is increasing evidence of improved outcomes with administration of higher doses (5–7). Before starting ERT for IOPD patients, the cross-reactive immunological material (CRIM), which is the presence of protein epitopes derived from acid alpha-glucosidase remnants in peripheral blood, needs to be ascertained as CRIM-negative individuals are at higher risk for developing sustained titers of anti-alglucosidase alpha neutralizing antibodies which can lead to treatment resistance and poorer outcomes (8). To prevent the development of anti-ERT antibodies, different immune tolerance induction protocols are used for CRIM-negative (9) and CRIM-positive (10) patients.
Sickle cell anemia (SCA, MIM #603903) is an autosomal recessive disorder of hemoglobin structure caused by the common HBB pathogenic variant (NM_000518.5):c.20A>T, p.(Glu7Val) which causes polymerization of deoxyhemoglobin, resulting in chronic intravascular hemolysis and intercurrent vaso-occlusive episodes. The chronic anemia of SCA requires cardiovascular adaptation with increased cardiac output to maintain tissue oxygen delivery, and individuals with SCA demonstrate increased stroke volume, left ventricle dilatation, and left ventricular hypertrophy starting in childhood and proportional to the severity of anemia (11, 12). Individuals with SCA also demonstrate decreased muscle microvascular oxygen delivery, decreased growth velocity, and lower muscle mass compared with healthy children (12, 13).
In this article, we report a patient diagnosed with IOPD and SCA. Although treated with ERT since the first month of life, his clinical course was complicated by left ventricular hypertrophy followed by early-onset dilation in infancy. This is a unique case where both conditions co-occurred in a patient with a severe clinical presentation and course.
Case report
A male newborn of African American ethnicity was born at 39 weeks gestational age from non-consanguineous parents, both previously diagnosed as heterozygous carriers for the HBB c.20A>T, p.(Glu7Val) variant. The pregnancy was otherwise uncomplicated. Shortly after birth, he developed respiratory distress requiring transient non-invasive respiratory support and admission to the neonatal intensive care unit. A chest radiograph exhibited marked cardiomegaly (Figure 1A) and physical exam was significant for hypotonia and mild macroglossia. An echocardiogram showed severe left ventricular hypertrophy (Figure 1B). Further studies revealed elevated pro B-type natriuretic peptide (pro-BNP) at 25,701 pg/mL (normal <10,000 pg/mL), creatine-kinase (CK) at 1,374 U/L (normal <300 U/L), and aldolase at 37.8 U/L (normal 0.1−8 U/L). Work-up for other metabolic and genetic diseases, including acylcarnitine profile, total and free carnitine, ammonia, lactate, serum amino acids, and urine organic acids, was non-contributory. Karyotype and chromosomal microarray did not detect abnormalities. A cardiomyopathy next-generation sequencing (NGS) panel analyzing the coding regions of 163 genes associated with cardiac disease was performed at the Washington University Genomic and Pathology Services. Two GAA variants, NM_000152.5: c.842G>C, p.(Arg281Pro) and NM_000152.5: c.2560C>T, p.(Arg854*) were found in compound heterozygosity. While the genetic panel was pending, newborn screening (NBS) showed a decreased GAA enzyme activity at 4% (normal >22%). The hemoglobinopathy portion of the NBS showed absence of Hb A and presence of Hb F and S, consistent with SCA. Confirmatory GAA enzyme activity was low at 0.8 pmol/punch/h (normal >3.88) and urine glucotetrasaccharide (Hex4) was elevated at 29.2 mmol/mol creatinine (normal <20) on day of life (DOL) 14. CRIM status determined by Western blotting was positive. The patient was started on alglucosidase alfa infusions (20 mg/kg every 2 weeks) with an immune tolerance induction protocol comprised of methotrexate at 0.4 mg/kg for three consecutive days, starting on the day prior to the infusion, on DOL 30. This protocol was used for the first three enzyme replacement therapy (ERT) infusions (total of 9 doses of methotrexate), as previously described (10).
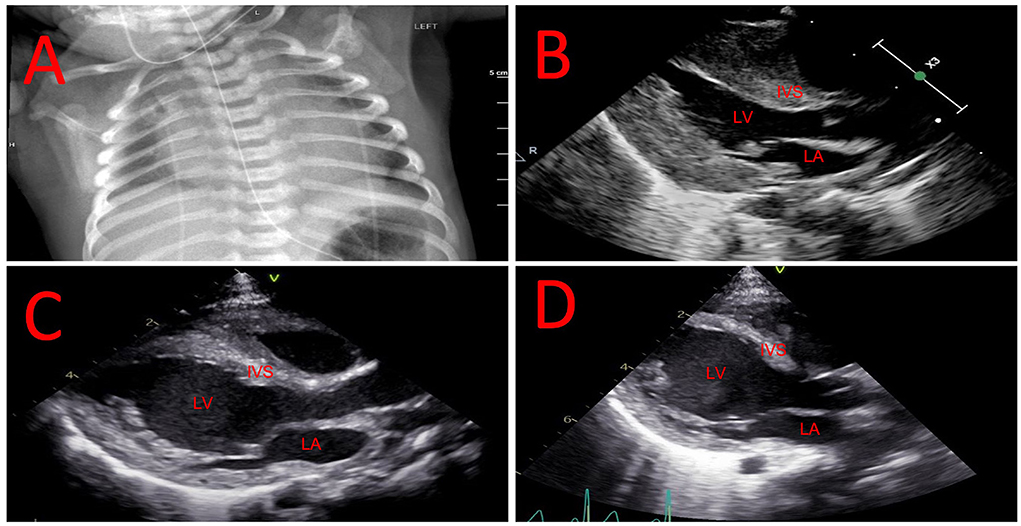
Figure 1. Evolution of cardiac hypertrophy and dilation. (A) Chest radiograph showing marked cardiomegaly in the neonatal period. (B) Echocardiogram showing hypertrophic left ventricle in the neonatal period. (C) Echocardiogram showing mixed left ventricular hypertrophy and dilation at 2 months of age. (D) Echocardiogram showing resolution of left ventricular hypertrophy with worsening of dilation at 7 months of age. IVS, interventricular septum; LA, left atrium; LV, left ventricle.
The SCA diagnosis was confirmed by HBB sequencing showing homozygosity for c.20A>T and negative HBB duplication/deletion analysis, ruling out hereditary persistence of fetal hemoglobin. He was started on penicillin V potassium prophylaxis at 1 month old. Due to concern that chronic anemia would adversely impact his cardiac function, and that any SCA vaso-occlusive complications such as acute chest syndrome or splenic sequestration in infancy would be poorly tolerated, chronic red blood cell transfusion therapy was chosen as his primary disease-modifying therapy. His first transfusion was administered at 2 months of age when Hb decreased to 8.4 g/dl. His transfusion therapy goal is to maintain Hb >9.5 g/dl to minimize the physiological stress of anemia. At 2 months of life, the patient was noticed to have new-onset left ventricular dilation in addition to cardiac hypertrophy (Figure 1C). Due to concerns for a decrease in left ventricular systolic function the patient was started on enalapril to prevent cardiac remodeling.
He has had 3 readmissions for respiratory decompensation due to viral infections: dual SARS-CoV-2 and respiratory syncytial virus infection at 3 months old (requiring high-flow nasal cannula), human metapneumovirus infection at 5 months old (requiring high-flow nasal cannula in the ICU setting) and rhino/enterovirus at 6 months old (requiring BiPAP in the ICU setting). At 7 months old, his outpatient ERT dosing was noted to having been based solely on birth weight, which resulted in a progressively decreasing ERT dose per kg body weight; on his most recently outpatient infusions this was ~13 mg/kg, and was corrected to 20 mg/kg. He was admitted subsequently at 7 months old due to respiratory failure with concern for an aspiration event requiring admission to the intensive care unit for non-invasive respiratory support. A repeat echocardiogram showed resolution of left ventricular hypertrophy with continuing dilation (Figure 1D). Evolution of systolic function and cardiac indices are presented in Figure 2. He has received a lifetime total of 10 pRBC transfusions at the time of this report, with a hemoglobin nadir of 7.7 g/dL. He remains gastrostomy-dependent. He continues to exhibit very low axial and appendicular muscle tone. He has required ~150–160 kcal/kg/day of enteral nutrition since birth for growth; non-etheless, he was at <1% (Z-score = −2.53, WHO Boys 0–2 years) for weight and <1% (Z-score = −3.57, WHO Boys 0–2 years) for weight-for-length at his most recent admission.
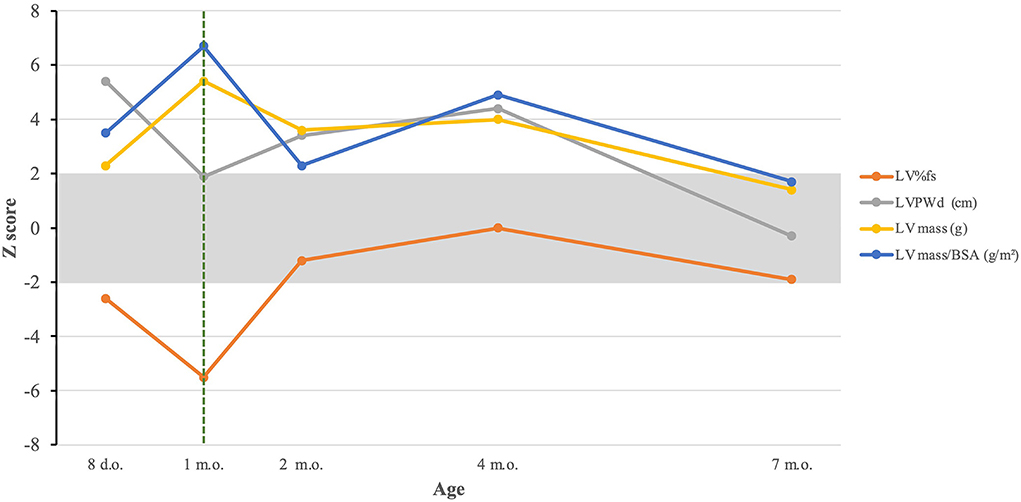
Figure 2. Evolution of cardiac indices in response to treatment. The dashed green line represents initiation of ERT. LV%fs, left ventricular percentual fractional shortening; LVPWd, left ventricular posterior wall dimension; LV mass, left ventricular mass; LV mass/BSA, left ventricular mass/body surface area.
Discussion
IOPD, with early onset and a lethal prognosis, can be modified through intervention and therefore meets generally accepted consensus criteria for inclusion in NBS programs (14). In the United States of America, the inclusion of Pompe disease in NBS was recommended in 2015, and currently more than 20 states have started the necessary implementation steps (15). The incidence of Pompe disease is historically reported as ~1:40,000 live births Western countries (16); however, this epidemiology has been calculated mainly based on European populations, with blind studies showing a higher incidence in other populations including people of African ancestry (17). Moreover, after implementation of universal screening through NBS there have been many reports of an incidence up to 4x higher than expected [as summarized by Davids et al. (18)], pointing to a substantial amount of historically missed diagnoses. SCA, also an autosomal recessive disorder included in NBS, contrasts with Pompe disease by having a high populational prevalence, with ~100,000 diagnosed Americans, of whom ~90% are of African ancestry (19). Prenatal genetic diagnosis for SCA and IOPD, as for most genetic disorders, is still incipient but may allow for even swifter initiation of treatment in future patients. In this article, we have presented a case of concomitant Pompe disease—presenting as IOPD—and SCA. The lack of previous reports of this co-incidence may reflect an epidemiological gap and probable underdiagnosis of Pompe disease in people of African ancestry, reinforcing previous studies (17) and underscoring the importance of universal NBS for equanimity in healthcare.
Two heterozygous GAA variants were detected. The p.(Arg854*) variant is predicted to result in a premature stop codon, nonsense-mediated decay, and has been demonstrated to result in an absent gene product (20); this variant has been observed in homozygous or compound heterozygous states in CRIM-negative individuals diagnosed with Pompe disease (21, 22). Published functional studies of the p.(Arg845*) variant have shown negligible activity compared to wild-type protein (23). This variant is commonly observed in individuals of African descent with IOPD (24, 25). This variant meets criteria PVS1, PM2, PM3, and PP4 of the American College of Medical Genetics/Association for Molecular Pathology (ACMG/AMP) guidelines for the interpretation of sequence variants (26) and is thus classified as pathogenic. We also identified a novel variant, c.842G>C, p.(Arg281Pro), in this patient, not previously observed in individuals with Pompe disease. A different amino acid change at the same codon, c.841C>T, p.(Arg281Trp), has been reported as a known likely pathogenic variant (27). Our patient's variant is absent in gnomAD, and multiple in silico prediction algorithms are in agreement that this variant may have a deleterious/probably damaging effect on protein function. Given the available evidence and in accordance with the ACMG/AMP guidelines, as well as the ClinGen GAA sequence analysis recommendations from ClinGen, the GAA p.(Arg281Pro) variant meets criteria PM2, PM3, PM5, PP3, and PP4 and was classified as likely pathogenic. Segregation studies on parental sample further confirmed that the p.(Arg845*) and p.(Arg281Pro) are in trans configuration, consistent with biallelic disruption of the gene product.
Treatment of IOPD with alglucosidase alfa ERT has been shown to improve survival, functionality, cardiomyopathy, and weight gain of individuals with IOPD (28), with the better outcomes observed in patients started on ERT in the first month of life (29), as was the case in our patient. In contrast with most other lysosomal diseases, the start of treatment in IOPD is contingent upon determination of CRIM status for appropriate immune tolerance induction as without immunomodulation, virtually all CRIM-negative patients develop a high sustained immune response to ERT that leads to dismal outcomes (9, 30). Although approximately half of CRIM-positive patients do not have significant antibody titers, 32–40% of CRIM-positive patients who receive ERT without immune tolerance induction can develop intermediate- or high sustained antibody titers, leading to worse outcomes when compared to CRIM-positive patients that received immunomodulating agents (8, 31). Currently, the validated immune tolerance induction regimen for CRIM-positive patients contains methotrexate (10), a potent myelotoxic folate antagonist that can lead to myelosuppression. Although this regimen utilizes low-dose, short-term methotrexate, which is generally welltolerated in IOPD patients, this may not be the most suitable regimen for a patient with concomitant bone marrow disease. Evidence for the use of methotrexate in patients with SCA is scarce and includes reports of transient cytopenia despite the concomitant administration of folinic acid (32). In this case report, anemia—which may have been exacerbated by the use of methotrexate—may have led to worsening cardiac dilatation despite ERT, highlighting the complexity of medical care in individuals with a dual diagnosis. As the incidence of Pompe disease increases as a result of universal NBS, more patients with concurrent IOPD and SCA (or other hemoglobinopathies) are expected to be diagnosed, and it becomes necessary to study novel immune tolerance induction protocols without the use of methotrexate, or with maximized support (with agents such as leucovorin or with preventive blood transfusions) for safe ERT initiation in this population. As newer recombinant enzymes and gene therapies are being tested, it will be important that ITI regimens are also validated for co-morbid populations.
Finally, decisions about SCA management were made based on knowledge that growth and cardiac function are more normal in children with SCA who have higher hemoglobin concentrations. Typically, chronic red blood cell transfusion therapy is utilized in SCA for stroke prevention with a primary goal of maintaining Hb S <30%, which may result in a nadir Hb pre-transfusion of <8 g/dl (33). In the current case, since this degree of anemia may compromise growth and increase the risk of cardiac stress, a transfusion strategy similar to that utilized in children with beta-thalassemia major was chosen, in which total hemoglobin concentration is maintained at 9–10 g/dl to minimize anemia-related organ dysfunction (34). Chronic transfusions will cause iron overload, which over many years can increase risk of heart failure but is manageable with iron chelating drugs (34). Hydroxyurea is another option for disease-modifying therapy, but it is not recommended until age 9 months. Patients with SCA have varying response to hydroxyurea, but in general the Hb remains higher on chronic red blood cell transfusion therapy than on this medication (33). Converting to hydroxyurea therapy from transfusion therapy may be a possibility if cardiac function remains stable and his strength is improving as he gets older, with careful monitoring of cardiac and skeletal muscle function. Hematopoietic stem cell transplantation can be curative for sickle cell disease; however, it has a delicate risk/benefit balance and requires a good functional status, thus not being a feasible option for this patient.
Conclusion
We have presented a unique case of concomitant IOPD and SCA leading to complex initiation of ERT and need for aggressive transfusion support to prevent cardiomyopathy due to anemia. As more locations implement NBS for Pompe disease, it is expected that such cases will become more common, and novel protocols that take the myelotoxicity of immune tolerance induction regimens into account will become necessary.
Data availability statement
The original contributions presented in the study are included in the article/supplementary materials, further inquiries can be directed to the corresponding author.
Ethics statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author contributions
RS wrote the first version of the manuscript. RS, PS, JG, and PD saw the patient at presentation, made the clinical and biochemical diagnosis of Pompe disease and started immune tolerance induction, and enzyme replacement therapy. RS, KL, and HN continued to follow the patient at the genetic clinic appointments, infusions, and admissions. LM saw the patient at follow-up and counseled the family. Y-CH and MS made the molecular diagnosis and wrote the variant analysis. MH made the diagnosis of sickle cell anemia, the decision for chronic transfusion therapy, and has been following the patient throughout life. JC made all the cardiological diagnoses and has been following the patient throughout life. LV and HW have been following the patient from a critical care perspective throughout life. All other authors provided feedback on the manuscript, reviewed it, and approved the final version and the decision to submit it for publication.
Funding
Funds for publication were provided to PD by Washington University in Saint Louis.
Acknowledgments
The authors are grateful for the patient's family for the trust, as well as to the staff of Saint Louis Children's Hospital, especially Dr. Karolina Kata for the remarkable care coordination during the first month of life.
Conflict of interest
Author PD declares the following conflicts of interest: Genzyme: research support, M6P Therapeutics: research support, and Mandos Health: consulting. Author MH declares the following conflict of interest: Pfizer, Inc: spouse employment, Bluebird Bio: consulting, Forma Therapeutics: research funding, Global Blood Therapeutics: research funding and consulting.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D, et al. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. (2006) 148:671–6. doi: 10.1016/j.jpeds.2005.11.033
2. Gupta N, Kazi ZB, Nampoothiri S, Jadgeesh S, Kabra M, Puri RB, et al. Clinical and molecular disease spectrum and outcomes in patients with infantile-onset pompe disease. J Pediatr. (2020) 216:44–50.e5. doi: 10.1016/j.jpeds.2019.08.058
3. Chan J, Desai AK, Kazi ZB, Corey K, Austin S, Hobson-Webb LD, et al. The emerging phenotype of late-onset Pompe disease: a systematic literature review. Mol Genet Metab. (2017) 120:163–72. doi: 10.1016/j.ymgme.2016.12.004
4. Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL, et al. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. (2007) 68:99–109. doi: 10.1212/01.wnl.0000251268.41188.04
5. Poelman E, van den Dorpel JJA, Hoogeveen-Westerveld M, van den Hout JMP, van der Giessen LJ, van der Beek NAME, et al. Effects of higher and more frequent dosing of alglucosidase alfa and immunomodulation on long-term clinical outcome of classic infantile Pompe patients. J Inherit Metab Dis. (2020) 43:1243–53. doi: 10.1002/jimd.12268
6. van Gelder CM, Poelman E, Plug I, Hoogeveen-Westerveld M, van der Beek NAME, Reuser AJJ, et al. Effects of a higher dose of alglucosidase alfa on ventilator-free survival and motor outcome in classic infantile Pompe disease: an open-label single-center study. J Inherit Metab Dis. (2016) 39:383–90. doi: 10.1007/s10545-015-9912-y
7. de Las Heras J, Cano A, Vinuesa A, Montes M, Suarez UM, Arza A, et al. Importance of timely treatment initiation in infantile-onset pompe disease, a single-centre experience. Children (Basel). (2021) 8:11. doi: 10.3390/children8111026
8. Banugaria SG, Prater SN, Ng YK, Kobori JA, Finkel RS, Ladda RL, et al. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease. Genet Med. (2011) 13:729–36. doi: 10.1097/GIM.0b013e3182174703
9. Li C, Desai AK, Gupta P, Dempsey K, Bhambhani V, Hopkin RJ, et al. Transforming the clinical outcome in CRIM-negative infantile Pompe disease identified via newborn screening: the benefits of early treatment with enzyme replacement therapy and immune tolerance induction. Genet Med. (2021) 23:845–55. doi: 10.1038/s41436-020-01080-y
10. Kazi ZB, Desai AK, Troxler RB, Kronn D, Packman S, Sabbadini M, et al. An immune tolerance approach using transient low-dose methotrexate in the ERT-naïve setting of patients treated with a therapeutic protein: experience in infantile-onset Pompe disease. Genet Med. (2019) 21:887–95. doi: 10.1038/s41436-018-0270-7
11. Batra AS, Acherman RJ, Wong WY, Wood JC, Chan LS, Ramicone E, et al. Cardiac abnormalities in children with sickle cell anemia. Am J Hematol. (2002) 70:306–12. doi: 10.1002/ajh.10154
12. Gladwin MT, Sachdev V. Cardiovascular abnormalities in sickle cell disease. J Am Coll Cardiol. (2012) 59:1123–33. doi: 10.1016/j.jacc.2011.10.900
13. Charlot K, Antoine-Jonville S, Moeckesch B, Jumet S, Romana M, Waltz X, et al. Cerebral and muscle microvascular oxygenation in children with sickle cell disease: Influence of hematology, hemorheology and vasomotion. Blood Cells Mol Dis. (2017) 65:23–8. doi: 10.1016/j.bcmd.2017.03.015
14. Forman J, Coyle F, Levy-Fisch J, Roberts P, Terry S, Legge M. Screening criteria: the need to deal with new developments and ethical issues in newborn metabolic screening. J Community Genet. (2013) 4:59–67. doi: 10.1007/s12687-012-0118-9
15. Ames EG, Fisher R, Kleyn M, Ahmad A. Current Practices for U.S. Newborn Screening of Pompe Disease and MPSI. Int J Neonatal Screen. (2020) 6:72. doi: 10.3390/ijns6030072
16. Martiniuk F, Chen A, Mack A, Arvanitopoulos E, Chen Y, Rom WN, et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am J Med Genet. (1998) 79:69–72.
17. Limgala RP, Furtak V, Ivanova MM, Changsila E, Wilks F, Fidelia-Lambert MN, et al. Selective screening for lysosomal storage disorders in a large cohort of minorities of African descent shows high prevalence rates and novel variants. JIMD Rep. (2021) 59:60–8. doi: 10.1002/jmd2.12201
18. Davids L, Sun Y, Moore RH, Lisi E, Wittenauer A, Wilcox WR, et al. Health care practitioners' experience-based opinions on providing care after a positive newborn screen for Pompe disease. Mol Genet Metab. (2021) 134:20–8. doi: 10.1016/j.ymgme.2021.09.001
20. Hermans MM, van Leenen D, Kroos MA, Beesley CE, van der Ploeg AT, Sakuraba H, et al. Twenty-two novel mutations in the lysosomal alpha-glucosidase gene (GAA) underscore the genotype-phenotype correlation in glycogen storage disease type II. Hum Mutat. (2004) 23:47–56. doi: 10.1002/humu.10286
21. Abbott MA, Prater SN, Banugaria SG, Richards SM, Young SP, Rosenberg AS, et al. Atypical immunologic response in a patient with CRIM-negative Pompe disease. Mol Genet Metab. (2011) 104:583–6. doi: 10.1016/j.ymgme.2011.08.003
22. Bali DS, Goldstein JL, Banugaria S, Dai J, Mackey J, Rehder C, et al. Predicting cross-reactive immunological material (CRIM) status in Pompe disease using GAA mutations: lessons learned from 10 years of clinical laboratory testing experience. Am J Med Genet C Semin Med Genet. (2012) 160C:40–9. doi: 10.1002/ajmg.c.31319
23. Hermans MM, de Graaff E, Kroos MA, Wisselaar HÁ, Willemsen R, Oostra BA, et al. The conservative substitution Asp-645–>Glu in lysosomal alpha-glucosidase affects transport and phosphorylation of the enzyme in an adult patient with glycogen-storage disease type II. Biochem J. (1993) 289:687–93. doi: 10.1042/bj2890687
24. Becker JA, Vlach J, Raben N, Nagaraju K, Adams EM, Hermans MM, et al. The African origin of the common mutation in African American patients with glycogen-storage disease type II. Am J Hum Genet. (1998) 62:991–4. doi: 10.1086/301788
25. Kroos M, Hoogeveen-Westerveld M, van der Ploeg A, Reuser AJ. The genotype-phenotype correlation in Pompe disease. Am J Med Genet C Semin Med Genet. (2012) 160C:59–68. doi: 10.1002/ajmg.c.31318
26. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
27. Wittmann J, Karg E, Turi S, Legnini G, Giese AK, Lukas J, et al. Newborn screening for lysosomal storage disorders in Hungary. JIMD Rep. (2012) 6:117–25. doi: 10.1007/8904_2012_130
28. Nicolino M, Byrne B, Wraith JE, Leslie N, Mandel H, Freyer DR, et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med. (2009) 11:210–9. doi: 10.1097/GIM.0b013e31819d0996
29. Chien YH, Lee NC, Chen CA, Tsai FJ, Tsai WH, Shie JY, et al. Long-term prognosis of patients with infantile-onset Pompe disease diagnosed by newborn screening and treated since birth. J Pediatr. (2015) 166:985–91. doi: 10.1016/j.jpeds.2014.10.068
30. Desai AK, Baloh CH, Sleasman JW, Rosenberg AS, Kishnani PS. Benefits of prophylactic short-course immune tolerance induction in patients with infantile pompe disease: demonstration of long-term safety and efficacy in an expanded cohort. Front Immunol. (2020) 11:1727. doi: 10.3389/fimmu.2020.01727
31. Desai AK, Kazi ZB, Bali DS, Kishnani PS. Characterization of immune response in cross-reactive immunological material (CRIM)-positive infantile pompe disease patients treated with enzyme replacement therapy. Mol Genet Metab Rep. (2019) 20:100475. doi: 10.1016/j.ymgmr.2019.100475
32. Brandalise SR, Assis R, Laranjeira ABA, Yunes JA, de Campos-Lima PO. Low-dose methotrexate in sickle-cell disease: a pilot study with rationale borrowed from rheumatoid arthritis. Exp Hematol Oncol. (2017) 6:18. doi: 10.1186/s40164-017-0078-1
33. Ware RE, Helms RW, Investigators S. Stroke with transfusions changing to hydroxyurea (SWiTCH). Blood. (2012) 119:3925–32. doi: 10.1182/blood-2011-11-392340
Keywords: glycogen storage disorder type II, alpha-glucosidase, sickle cell anemia, immune tolerance induction, enzyme replacement therapy, methotrexate, newborn screening
Citation: Starosta RT, Hou Y-CC, Leestma K, Singh P, Viehl L, Manwaring L, Granadillo JL, Schroeder MC, Colombo JN, Whitehead H, Dickson PI, Hulbert ML and Nguyen HT (2022) Infantile-onset Pompe disease complicated by sickle cell anemia: Case report and management considerations. Front. Pediatr. 10:944178. doi: 10.3389/fped.2022.944178
Received: 14 May 2022; Accepted: 01 August 2022;
Published: 28 September 2022.
Edited by:
Shyi-Jou Chen, National Defense Medical Center, TaiwanReviewed by:
Yuan Gao, Shandong University, ChinaDer-Shiun Wang, Tri-Service General Hospital, Taiwan
Yohei Sato, Jikei University School of Medicine, Japan
Copyright © 2022 Starosta, Hou, Leestma, Singh, Viehl, Manwaring, Granadillo, Schroeder, Colombo, Whitehead, Dickson, Hulbert and Nguyen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rodrigo Tzovenos Starosta, ci5zdGFyb3N0YUB3dXN0bC5lZHU=