 Dongmei Liu1,2,3Jiali Yu1,2,3Xin Wang4Yang Yang1,2,3Li Yu1,2,3
Dongmei Liu1,2,3Jiali Yu1,2,3Xin Wang4Yang Yang1,2,3Li Yu1,2,3 Shi Zeng1,2,3Ming Zhang1,2,3
Shi Zeng1,2,3Ming Zhang1,2,3 Ganqiong Xu1,2,3*
Ganqiong Xu1,2,3*- 1Department of Ultrasound Diagnostic, The Second Xiangya Hospital, Central South University, Changsha, China
- 2Research Center of Ultrasound Diagnostic, The Second Xiangya Hospital, Central South University, Changsha, China
- 3Clinical Research Center for Medical Imaging in Hunan Province, Changsha, China
- 4Department of Obstetrics and Gynecology Prenatal Diagnosis Center, The Second Xiangya Hospital, Central South University, Changsha, China
Nemaline myopathy (NM) is a rare, hereditary heterogeneous myopathy. Fetal NM has a more severe disease course and a poorer prognosis and is usually lethal during the first few months of life. Hence, early prenatal diagnosis is especially important for clinical interventions and patient counseling. We report the case of a fetus with NM due to KLHL40 gene variation leading to arthrogryposis multiplex congenita (AMC). The ultrasonography and histopathology results revealed an enhanced echo intensity and decreased muscle thickness, which may be novel features providing early clues for the prenatal diagnosis of NM. Moreover, to our knowledge, this article is the first report to describe a case of NM associated with complex congenital heart disease (CHD).
Introduction
Nemaline myopathy (NM) is a rare congenital disease of skeletal muscle causing severe muscle weakness and other neuromuscular dysfunction manifestations. Shy first described it in 1963 (1) and Kaspersky first reported it as a prenatal diagnosis in 2008 (2). The incidence of NM is approximately 1:50,000 (3). Based on the time of onset and severity of symptoms, it can be classified into six forms ranging from severe congenital/neonatal onset to adult onset (4). The severe congenital subgroup accounts for 15% of all NM cases (5). NM is generally considered a hereditary disease, but its inheritance is heterogeneous, either dominant or recessive, and many loci are involved. Thus, NM causes various diseases. Variations in at least 12 genes including ACTA1, CFL2, KBTBD13, KLHL40, KLHL41, LMOD3, MYPN, NEB, TNNT1, TNNT3, TPM2, and TPM3, have been identified to be associated with the condition (6). Variations in the KLHL40 gene have been recently identified as a cause of severe autosomal-recessive NM. The diagnosis of NM is mainly based on genetic and pathological features. The characteristic pathological change is the deposition of a large number of nemaline bodies in muscle fibers. Prenatal ultrasound diagnosis of NM is difficult and thus rarely performed. This study aimed to describe novel ultrasound findings for the lethal form of NM. Enhanced echo intensity and decreased muscle thickness demonstrate that amyoplasia seems to be an element of the poor prognosis of these patients. Thus, when ultrasonographic features such as polyhydramnios, decreased fetal movements and club feet are noted during pregnancy, the echo and thickness of the muscle should be observed carefully to determine the potential existence of congenital neuromuscular disorders such as NM.
Case Report
A 36-year-old Chinese woman, gravida 1, para 0, was referred to our hospital at 36 weeks of gestation because of multiple cardiac anomalies (right ventricular double outlet and aortic coarctation) and bilateral foot inversion observed in the fetus. At 18 weeks gestation, amniocentesis showed that the fetus was at high risk for trisomy 18 and trisomy 21. The chromosomal microarray of amniocytes was normal. No exposure to teratogenic substances or family history of congenital malformation was noted. The pregnancy was uncomplicated.
The first ultrasound examination conducted in our department showed polyhydramnios, and the fetus was in breech presentation with no obvious limb movements, bilateral clubfoot, or tilted fingers. The cardiac malformation was the same as before. Interestingly, we found that echoes of the skeletal muscles were significantly enhanced (close to or slightly weaker than the bone echo) and had posterior echo attenuation (Figures 1A,B). Serial scans were performed the following week, which confirmed limb stiffness (Figures 2B,C). In addition, it was found by comparison that limb muscles of this fetus were thinner than those in the control. Ultrasonographic findings led to the suspicion of AMC associated with amyoplasia, complex congenital heart disease (CHD) (Figures 2E,F), and polyhydramnios.
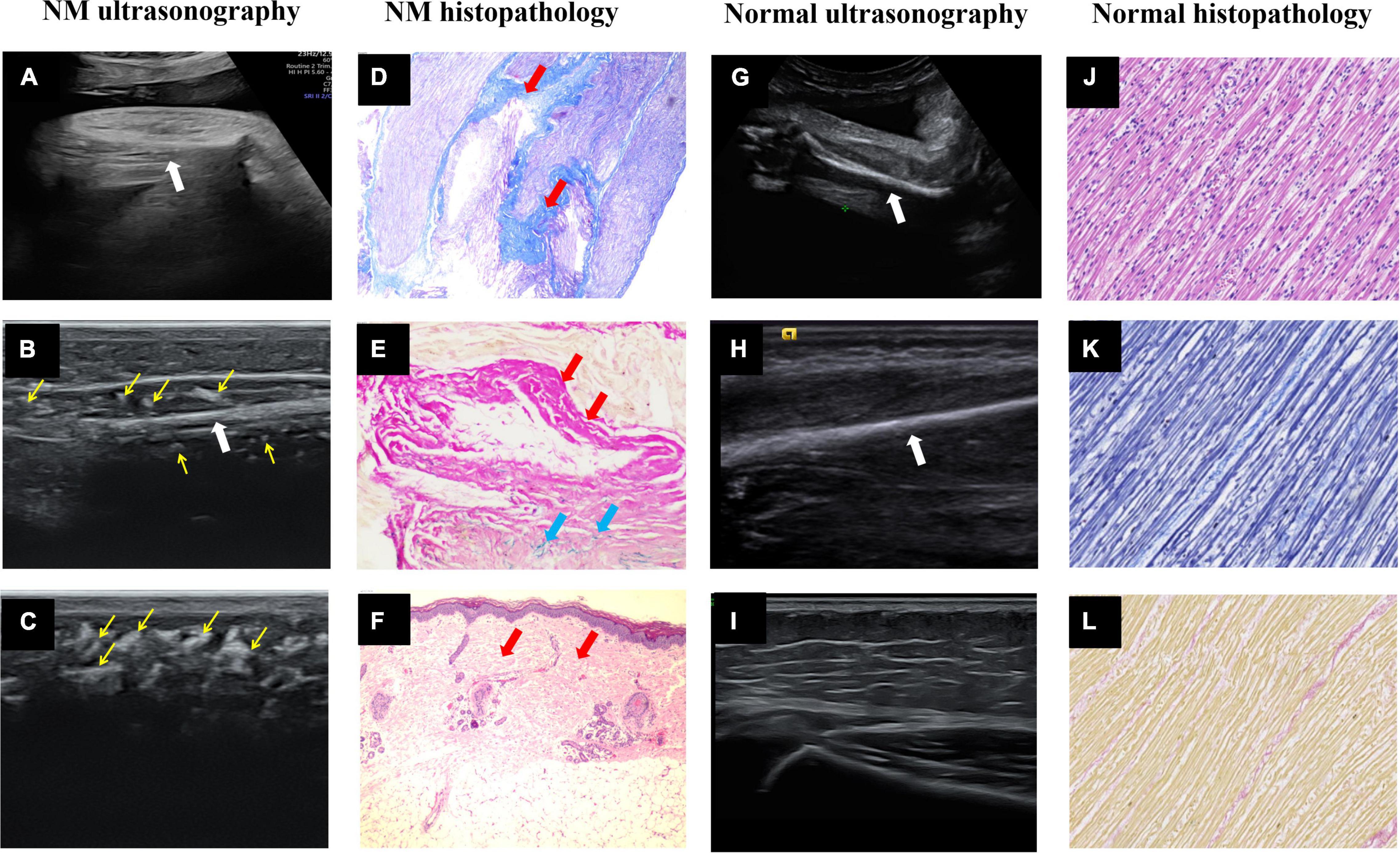
Figure 1. Ultrasound images and histopathology of the prenatal and postnatal extremities. (A) Intrauterine ultrasound examination. (B,C) Postpartum high-frequency probe examination showed NM fetal upper limb muscle echo close to the humerus echo, and many hyperechoic regions (thin yellow arrows) in the muscle layer and subcutaneous tissue. (D) Right calf muscle Masson trichrome staining × 40 and (E) right calf muscle elastic fiber staining × 200. The muscle fiber fascicles were sparse and replaced by collagen fibers (thick red arrows) and elastic fibers (thick blue arrows). (F) Right calf skin H&E × 40 showed fibrous tissue (thick red arrows) proliferated in the dermis layer and subcutaneous tissue. (G–I) The normal fetal upper limb muscle echo was significantly lower than the humerus echo, and the subcutaneous tissue had low echo intensity with several echogenic septa of connective tissue. The thick white arrows point to the long bones of the fetal limbs. (J–L) The normal fetal right calf muscle H&E × 40, Masson trichrome staining × 40 and elastic fiber staining ×40.
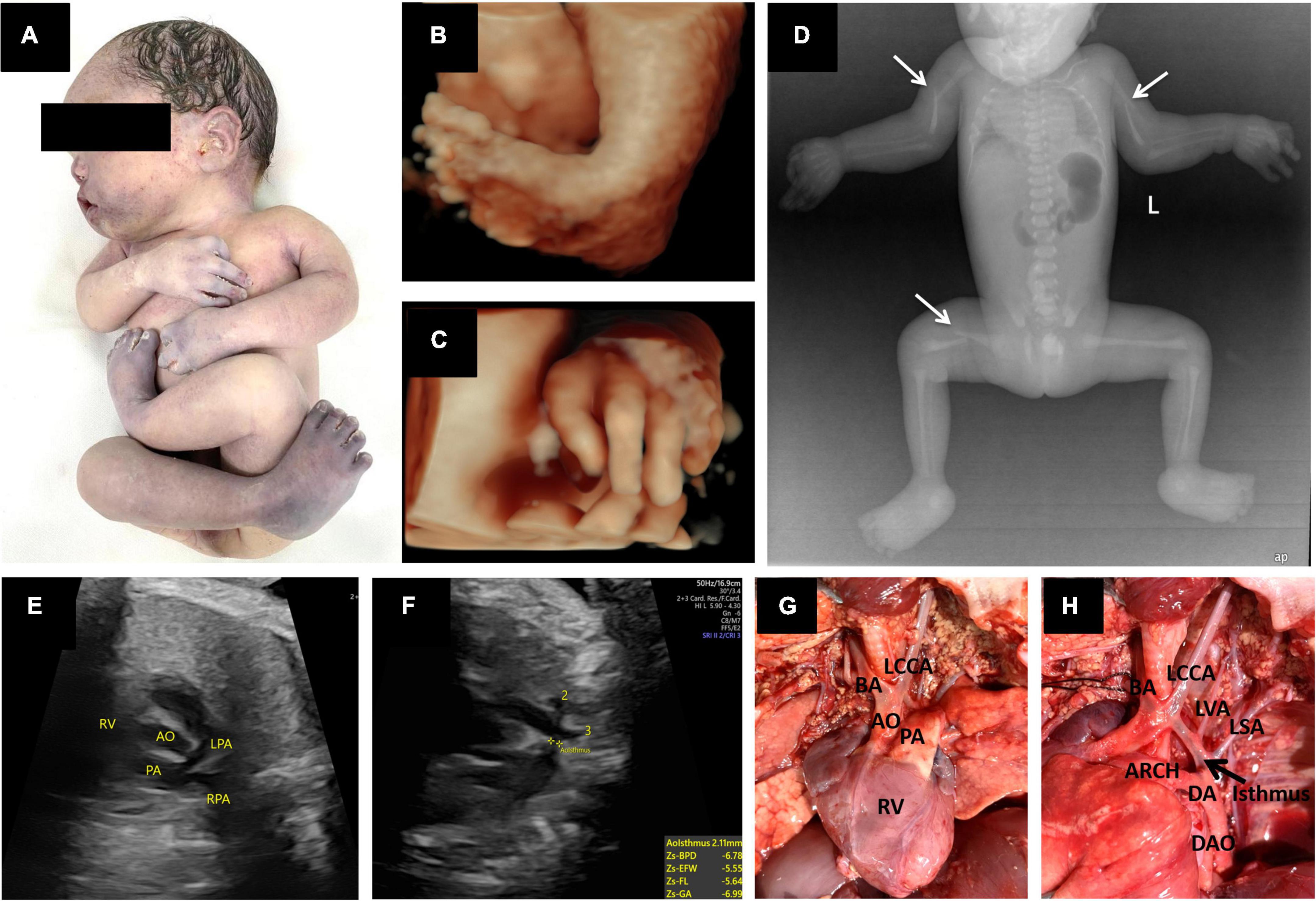
Figure 2. Prenatal 3D ultrasound images and fetal echocardiography, postpartum gross anatomical specimens, X-ray photographs and cardiac anatomy. (A) Postmortem photo demonstrated multiple contractures of the upper and lower extremities. (B,C) Three-dimensional ultrasound showed fetal foot inversion and tilted fingers. (D) X-rays showed bilateral humerus and right femur fractures (thin white arrow). (E,F) Prenatal echocardiography showed a right ventricular double outlet with coarctation of the aortic arch. (G,H) Postpartum cardiac anatomy confirmed sonographic findings. In addition, there were four arteries of the aortic arch, from right to left: BA, the brachiocephalic artery; LCCA, the left common carotid artery; LVA, the left vertebral artery; and LSA, the left subclavian artery. RV, right ventricle; AO, aorta; PA, pulmonary artery; LPA, left pulmonary artery; RPA, right pulmonary artery; DA, ductus arteriosus; DAO, descending aorta; ARCH, aortic arch.
Whole-exome sequencing (WES) was then performed on fetal umbilical cord blood and revealed a homozygous variation in the KLHL40 gene, NM_152393.2:c.1516A > C (p. Thr506Pro). Sanger sequencing verified that the patient was homozygous variation for c.1516A > C (p. Thr506Pro) (Supplementary Figure 1). According to ACMG guidelines, the variant was classified as pathogenic (PM1 + PM2 + PM3 + PP1 + PP4).
The pregnancy was selectively terminated after the patient received counseling. Postmortem radiological examination, necropsy (Figures 2A,D), and pathology confirmed all sonographic findings and revealed other malformations. X-rays showed pathological fractures. The autopsy revealed that the skin had increased hardness and decreased elasticity. The subcutaneous fat layer was significantly thickened, and the muscle layer was significantly thinner (Figures 3A,B). In addition, there were four arteries in the aortic arch (Figures 2G,H).
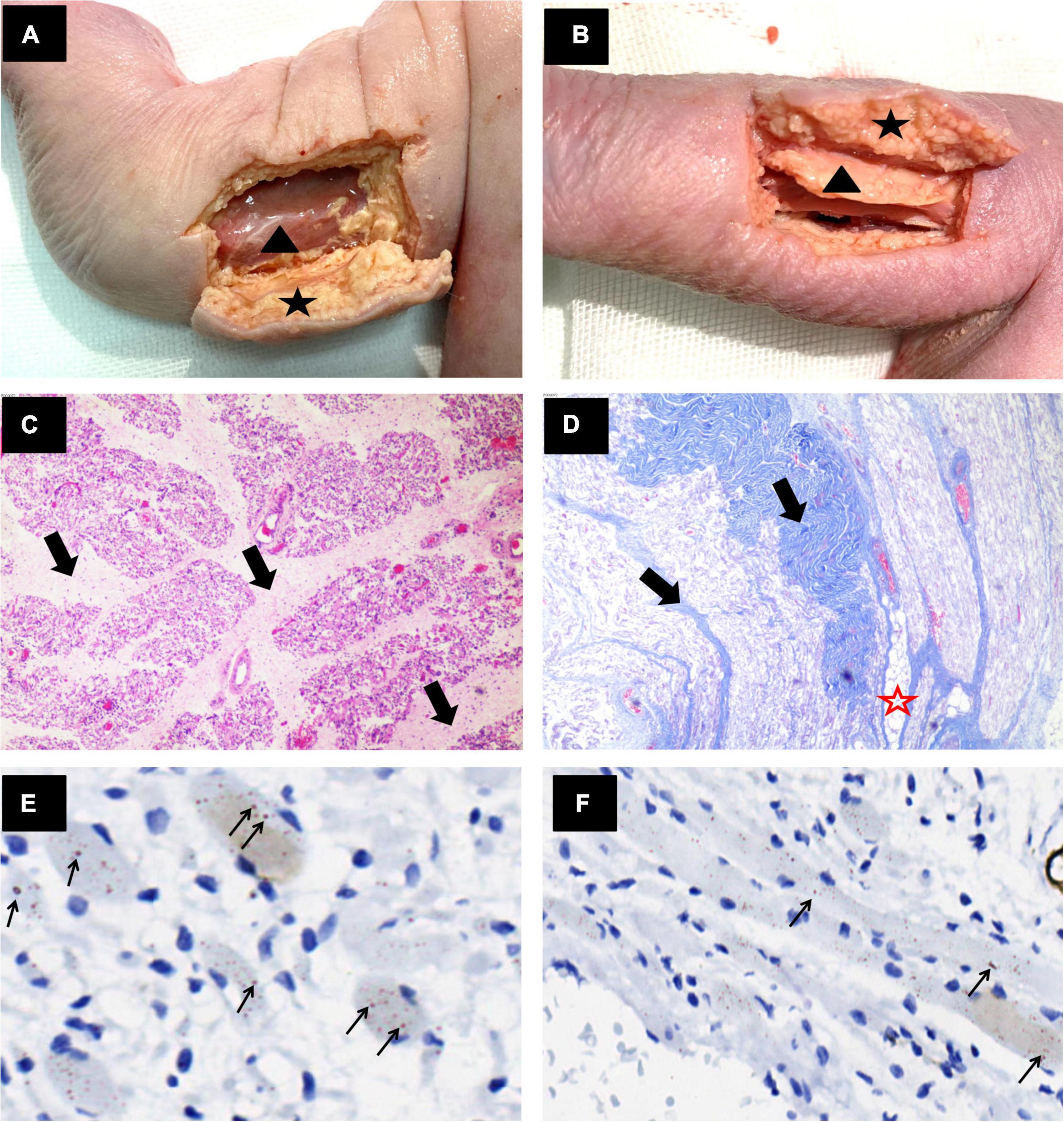
Figure 3. Anatomical and pathological images of the limb muscles. (A,B) Gross anatomy showed thickened subcutaneous fat (black star) and a thin muscle layer (black triangle) in the lower limbs of the fetus. (C) Right thigh muscle cross section H&E × 40 and (D) Right forearm muscle longitudinal section Masson trichrome × 40. The muscle bundles showed marked variation in size, and muscle fibers were replaced by fibrous tissue (thick black arrows) and adipose tissue (red star). (E,F) Right calf muscle cross section and longitudinal section anti-α-actinin antibody stained × 400. The muscle fibers were sparse, degenerated, and atrophied. Lots of nemaline rods in muscle fibers were strongly stained with anti-α-actinin antibody (thin black arrow).
Histopathological examination showed the following: (1) The muscle fibers were sparse, degenerated, and atrophied and were replaced by fatty-fibers (Figures 3C–F). (2) The dermis layer and subcutaneous tissue were thickened with fibrous tissue proliferation (Figure 1F); and (3) Nemaline bodies were positively stained with the anti-α-actinin antibody (Figures 3E,F). Unfortunately, we failed to perform electron microscopy. Altogether, the results of these examinations indicated a diagnosis of nemaline myopathy due to KLHL40 gene variation leading to fetal AMC associated with CHD.
Discussion
AMC is a heterogeneous disease characterized by multiple congenital joint contractures. This is the result of reduced fetal movement during development. AMC has been shown to be associated with more than 400 diseases and 350 known genes. AMC has been reported in association with neuromuscular disorders. NM is one of the congenital myopathies that leads to AMC (7). Fetal NM is a severe form of NM that carries a poor prognosis. Fetal NM due to variated KLHL40 is one of the most severe subgroups characterized by fetal akinesia (8), contractures, severe muscle weakness, respiratory failure, dysphagia, and early perinatal death (average age of death is 5 months) (9, 10). Currently, there is no available cure for fetal NM, and it has an extremely poor prognosis. Thus, early prenatal diagnosis is especially important. Several studies have suggested that prenatal identification of NM is challenging and can only be achieved by WES or fetal muscle biopsy. However, less than half of the muscle histology results of suspected NM cases present with nemaline bodies (5, 11), and WES cannot be performed for all fetuses. Therefore, prenatal ultrasound is one of the best indicators of the need to perform further WES analyses of affected fetuses.
To date, 34 prenatally diagnosed cases of NM have been reported in the literature (Table 1). Decreased fetal movement (74%), polyhydramnios (65%), and arthrogryposis (47%) were common prenatal features on sonography. Other less frequent findings included fetal edema (32%), clubfoot (26%), facial abnormalities (15%), lung hypoplasia (6%), and fractures (3%). Notably, our study is the first to describe the features of enhanced echo intensity and decreased thickness of skeletal muscle. In normal fetuses, muscle boundaries are clearly visible and the skeletal muscle echo is lower than or close to the liver echo. However, this fetus appeared to have unclear muscle boundaries. The muscle layer appeared thinned and had an enhanced echo (stronger than the liver echo and slightly weaker than or even approaching the bone echo, Figures 1A,G). Then, we used a high-frequency probe to compare the skin and muscle of this fetus to that of a normal infant (Figures 1B,C,H,I). Strikingly, the fetus demonstrated replacement of myofibers with connective tissue on ultrasonography (many hyperechoic regions in the muscles) and histopathology (Figures 1D,E). These were in stark contrast to the muscles of normal fetus (Figures 1J–L). Moreover, fibrous tissue proliferated in the dermis layer and subcutaneous tissue (Figure 1F), which exhibited several hyperechoic regions, replacing the several echogenic septa observed on ultrasonography (Figures 1C,I). Increased interstitial connective tissue is indicative of myopathy (12). In this case, the echo intensity increased because the muscular architecture was disrupted by muscle cell replacement with connective and fat tissue. These antenatal signs demonstrated that amyoplasia seemed to contribute to the poor prognosis. We recommend that decreased fetal movements combined with polyhydramnios should be used to observe the fetal joint activity and the echo and thickness of the skeletal muscle. Improving awareness and identification of abnormal ultrasound manifestations early alerts clinicians of the potential existence of neuromuscular disorders such as NM.

Table 1. Summary of prenatal ultrasound characteristics of nemaline myopathy.
NM needs to be differentiated from other neuromuscular disease, such as congenital muscular dystrophy (13). It can also show decreased fetal movement, polyhydramnios, arthrogryposis on prenatal ultrasound. To the best of our knowledge, whether the enhanced echo intensity and decreased thickness of skeletal muscle also appear has not been reported in the literature. In the future, we will investigate whether other congenital myopathies also demonstrate this abnormal ultrasound performance. Congenital muscular dystrophy is associated with muscle enzymes significantly increased over 10 times, and some have white matter lesions (14). In addition, polymyositis, dermatomyositis, and mitochondrial encephalopathy also need to be distinguished from NM. Polymyositis is rare in the neonatal period. Dermatomyositis often has obvious skin lesions (15). Mitochondrial encephalomyopathy is a multisystem disease mainly involving the brain and muscle (16). Diagnosis can be further confirmed by histopathology. Congenital muscular dystrophy also show histological loss of muscle fibers and the replacement of muscle tissue by connective and adipose tissue (13). However, typical nemaline rods are characteristic of NM. Staining with Gomori trichrome or electron microscopy can substantiate the nemaline rods (17). Ultimately, WES provides a precise genetic diagnosis.
One limitation of our study was that electron microscopy was not performed because fresh specimens were not available. Another limitation is the small number of cases. We will evaluate more NM cases in the future to study the correlation between ultrasonography features and unfavorable outcomes.
Conclusion
The large size and complexity of the genome necessitates a costly and lengthy determination of presence of neuromuscular disorders. Therefore, early abnormal ultrasound findings (decreased fetal movements, polyhydramnios, enhanced echo intensity, and decreased muscle thickness) may prompt clinicians to suspect congenital neuromuscular disease. It may provide early clue of targeted genes to facilitate a prenatal diagnosis of NM, followed by WES and biopsy to confirm the diagnosis. This is important to offer parents informed choices regarding the subsequent course of the pregnancy and to alert physicians to plan appropriate investigations. Furthermore, NM can be associated with CHD. To the best of our knowledge, this is the first report of NM associated with complex CHD.
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Medical Ethics Committee, the Second Xiangya Hospital, Central South University. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
DL and GX contributed to conception and design of the study. DL completed the data collection and wrote the first draft of the manuscript. DL, JY, XW, YY, LY, SZ, and MZ wrote sections of the manuscript. All authors contributed to manuscript revision and read and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81801721) and the Natural Science Foundation of Hunan Province (No. 2019JJ50880).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.937668/full#supplementary-material
Supplementary Figure 1 | Variant identification by Sanger sequencing. The red arrow represented the variant site (c.1516A > C).
References
1. Shy GM, Engel WK, Somers JE, Wanko T. Nemaline myopathy. A new congenital myopathy. Brain. (1963) 86:793–810.
2. Kasperski SB, Brennan AM, Corteville JE, Finkel RS, Golden J, Johnson MP, et al. Utility of fetal muscle biopsy for diagnosis of nemaline myopathy. Fetal Diagn Ther. (2008) 24:400–4. doi: 10.1159/000170095
3. North KN, Laing NG, Wallgren-Pettersson C. Nemaline myopathy: current concepts. The ENMC International consortium and nemaline myopathy. J Med Genet. (1997) 34:705–13. doi: 10.1136/jmg.34.9.705
4. Lehtokari V-L, Kiiski K, Sandaradura SA, Laporte J, Repo P, Frey JA, et al. Mutation update: the spectra of nebulin variants and associated myopathies. Hum Mutat. (2014) 35:1418–26. doi: 10.1002/humu.22693
5. Feingold-Zadok M, Chitayat D, Chong K, Injeyan M, Shannon P, Chapmann D, et al. Mutations in the NEB gene cause fetal akinesia/arthrogryposis multiplex congenita. Prenat Diagn. (2017) 37:144–50. doi: 10.1002/pd.4977
6. Sewry CA, Laitila JM, Wallgren-Pettersson C. Nemaline myopathies: a current view. J Muscle Res Cell Motil. (2019) 40:111–26. doi: 10.1007/s10974-019-09519-9
7. Vardon D, Chau C, Sigodi S, Figarella-Branger D, Boubli L. Congenital rapidly fatal form of nemaline myopathy with fetal hydrops and arthrogryposis. A case report and review. Fetal Diagn Ther. (1998) 13:244–9. doi: 10.1159/000020847
8. Chen T-H, Tian X, Kuo P-L, Pan H-P, Wong L-JC, Jong Y-J. Identification of KLHL40 mutations by targeted next-generation sequencing facilitated a prenatal diagnosis in a family with three consecutive affected fetuses with fetal akinesia deformation sequence. Prenat Diagn. (2016) 36:1135–8. doi: 10.1002/pd.4949
9. Yi S, Zhang Y, Qin Z, Yi S, Zheng H, Luo J, et al. A novel and recurrent KLHL40 pathogenic variants in a Chinese family of multiple affected neonates with nemaline myopathy 8. Mol Genet Genomic Med. (2021) 9:e1683. doi: 10.1002/mgg3.1683
10. Avasthi KK, Agarwal S, Panigrahi I. Mutation associated with severe nemaline myopathy, fetal akinesia, and cleft palate. J Pediatr Neurosci. (2019) 14:222–4. doi: 10.4103/jpn.JPN_60_19
11. Malfatti E, Lehtokari VL, Bohm J, De Winter JM, Schaffer U, Estournet B, et al. Muscle histopathology in nebulin-related nemaline myopathy: ultrastrastructural findings correlated to disease severity and genotype. Acta Neuropathol Commun. (2014) 2:44. doi: 10.1186/2051-5960-2-44
12. Berkenstadt M, Pode-Shakked B, Barel O, Barash H, Achiron R, Gilboa Y, et al. LMOD3-associated nemaline myopathy: prenatal ultrasonographic, pathologic, and molecular findings. J Ultrasound Med. (2018) 37:1827–33. doi: 10.1002/jum.14520
13. Vuopala K, Leisti J, Herva R. Lethal arthrogryposis in Finland–a clinico-pathological study of 83 cases during thirteen years. Neuropediatrics. (1994) 25:308–15. doi: 10.1055/s-2008-1073045
14. Mary P, Servais L, Vialle R. Neuromuscular diseases: diagnosis and management. Orthop Traumatol Surg Res. (2018) 104:S89–95. doi: 10.1016/j.otsr.2017.04.019
15. Findlay AR, Goyal NA, Mozaffar T. An overview of polymyositis and dermatomyositis. Muscle Nerve. (2015) 51:638–56. doi: 10.1002/mus.24566
16. Scarpelli M, Todeschini A, Volonghi I, Padovani A, Filosto M. Mitochondrial diseases: advances and issues. Appl Clin Genet. (2017) 10:21–6. doi: 10.2147/TACG.S94267
17. Zhao B, Dai T, Zhao D, Ma X, Zhao C, Li L, et al. Clinicopathologic profiles of sporadic late-onset nemaline myopathy: practical importance of anti-α-actinin immunostaining. Neurol Neuroimmunol Neuroinflamm. (2022) 9:e1184. doi: 10.1212/NXI.0000000000001184
18. Vendittelli F, Manciet-Labarchède C, Gilbert-Dussardier B. Nemaline myopathy in the neonate: two case reports. Eur J Pediatr. (1996) 155:502–5.
19. Lammens M, Moerman P, Fryns JP, Lemmens F, van de Kamp GM, Goemans N, et al. Fetal akinesia sequence caused by nemaline myopathy. Neuropediatrics. (1997) 28:116–9. doi: 10.1055/s-2007-973683
20. Wallgren-Pettersson C, Donner K, Sewry C, Bijlsma E, Lammens M, Bushby K, et al. Mutations in the nebulin gene can cause severe congenital nemaline myopathy. Neuromuscul Disord. (2002) 12:674–9.
21. Kuwata T, Matsubara S, Ohkusa T, Yada Y, Suzuki M. Decreased fetal movement prompts investigation of prenatal/neonatal nemaline myopathy: the possible merit of fetal movement count. J Obstet Gynaecol Res. (2011) 37:921–5. doi: 10.1111/j.1447-0756.2010.01438.x
22. Lawlor MW, Ottenheijm CA, Lehtokari V-L, Cho K, Pelin K, Wallgren-Pettersson C, et al. Novel mutations in NEB cause abnormal nebulin expression and markedly impaired muscle force generation in severe nemaline myopathy. Skelet Muscle. (2011) 1:23. doi: 10.1186/2044-5040-1-23
23. Yonath H, Reznik-Wolf H, Berkenstadt M, Eisenberg-Barzilai S, Lehtokari V-L, Wallgren-Pettersson C, et al. Carrier state for the nebulin exon 55 deletion and abnormal prenatal ultrasound findings as potential signs of nemaline myopathy. Prenat Diagn. (2012) 32:70–4. doi: 10.1002/pd.2905
24. Ahmed AA, Skaria P, Safina NP, Thiffault I, Kats A, Taboada E, et al. Arthrogryposis and pterygia as lethal end manifestations of genetically defined congenital myopathies. Am J Med Genet A. (2018) 176:359–67. doi: 10.1002/ajmg.a.38577
25. Abbott M, Jain M, Pferdehirt R, Chen Y, Tran A, Duz MB, et al. Neonatal fractures as a presenting feature of LMOD3-associated congenital myopathy. Am J Med Genet A. (2017) 173:2789–94. doi: 10.1002/ajmg.a.38383
26. Wang Y, Zhu C, Du L, Li Q, Lin M-F, Férec C, et al. Compound heterozygosity for novel truncating variants in the gene as the cause of polyhydramnios in two successive fetuses. Front Genet. (2019) 10:835. doi: 10.3389/fgene.2019.00835
27. Yeung KS, Yu FNY, Fung CW, Wong S, Lee HHC, Fung STH, et al. The KLHL40 c.1516A>C is a Chinese-specific founder mutation causing nemaline myopathy 8: report of six patients with pre- and postnatal phenotypes. Mol Genet Genomic Med. (2020) 8:e1229. doi: 10.1002/mgg3.1229
28. Rocha ML, Dittmayer C, Uruha A, Korinth D, Chaoui R, Schlembach D, et al. A novel mutation in NEB causing foetal nemaline myopathy with arthrogryposis during early gestation. Neuromuscul Disord. (2021) 31:239–45. doi: 10.1016/j.nmd.2020.11.014
Keywords: nemaline myopathy, arthrogryposis multiplex congenita, KLHL40 gene, amyoplasia, prenatal diagnosis
Citation: Liu D, Yu J, Wang X, Yang Y, Yu L, Zeng S, Zhang M and Xu G (2022) Case Report: Prenatal Diagnosis of Nemaline Myopathy. Front. Pediatr. 10:937668. doi: 10.3389/fped.2022.937668
Received: 06 May 2022; Accepted: 22 June 2022;
Published: 19 July 2022.
Edited by:
Aleksandra Jezela-Stanek, National Institute of Tuberculosis and Lung Diseases, PolandReviewed by:
Martin Lammens, Antwerp University Hospital, BelgiumAnkur Singh, Banaras Hindu University, India
Copyright © 2022 Liu, Yu, Wang, Yang, Yu, Zeng, Zhang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ganqiong Xu, eHVnYW5xaW9uZ0Bjc3UuZWR1LmNu