 Ying Yu
Ying Yu Cuiyun Li
Cuiyun Li Wei Li1
Wei Li1 Jian Wang
Jian Wang Ruen Yao
Ruen Yao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 02 September 2022
Sec. Pediatric Neurology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.931667
This article is part of the Research Topic Insights in Pediatric Neurology: 2021 View all 20 articles
SATB1 variants causing developmental delay with dysmorphic facies and dental anomalies have been reported in a small cohort. Most patients present epilepsy as a main clinical feature in neurodevelopmental disorders; however, its treatment is unknown. Here, we present a Chinese patient with a de novo truncating variation in SATB1 who presented with mild developmental delay. We disclose the detailed anti-epileptic pharmacological treatment that enabled a favorable outcome. Our study provides important information that may aid clinicians in the prognosis and treatment of rare neurological developmental disorders caused by gene mutations.
Variants of SATB1 cause clinically overlapping but distinct neurodevelopmental disorders, including intellectual disability, muscle tone abnormalities, hypotonia, spasticity, epilepsy, behavioral problems, facial dysmorphisms, and dental abnormalities. Genotype-phenotype relationships associated with each pathophysiological mechanism have been identified (1). In the limited number of reported clinical cases, motor and speech delays have been the most prevalent neurological manifestations (92 and 89%, respectively). Epilepsy accounts for 61% of all reported nervous system-related phenotypes and is the only symptom with a relatively comprehensive pharmacological treatment. Studies on most neurodevelopmental disorders have proved that the control of early epilepsy is crucial for slowing the progression of neurological impairment and restoring normal neurological function (2, 3). Details of the specific epileptic condition and therapeutic interventions in patients are not available in existing reports of SATB1 variants, thus lacking the clinicians’ and patients’ perspectives of the treatment experience. This study reports a Chinese patient with a pathogenic SATB1 mutation manifested as epilepsy, growth retardation, and facial dysmorphisms. The treatment process of epilepsy and the growth and development history of the patient are elaborated. Our study provides relevant clinical and pharmacological information for the prognosis and treatment of neurological rare developmental disorders caused by gene mutations.
The proband was a 7-year-old girl. She was referred to the Medical Genetics Clinic of Shanghai Children’s Medical Center, Sanya Women and Children’s Hospital, presenting with global developmental delay. She was the only child of non-consanguineous Han Chinese parents. The patient had no abnormalities on prenatal examination and was born naturally at full term. Her parents were physically healthy and had no relevant family history.
The girl sought medical advice for the first time at age 2. She began to roll her eyes up and frequently stopped moving for several seconds, mostly on stimulation. In 2 months, she had a major seizure that lasted approximately 5 min, with convulsions affecting her whole body, her eyes rolling up, and her mouth foaming. Facial features were not recognizable, and dental abnormalities were not evident (Figure 1A). She was diagnosed with epilepsy at the local hospital. The EEG (Electroencephalography) showed sharp, sharp-slow, and spinous slow waves in the bilateral occipital areas, but were more prominent on the right side. There were more asynchronous, sharp, slow, and spike waves in the bilateral frontal, middle, and central regions. The patient started anti-epileptic drug therapy. Valproic acid was used for 2 months at a dose of 250 mg (25 mg/kg) twice daily. Seizures occurred several times during this period, and EEG did not improve significantly. For the following 9 months, the dose of valproic acid was reduced to 100 mg (10 mg/kg), and 80 mg oxcarbazepine (8 mg/kg) was introduced, both administered twice daily for better control of the epileptic activity. With the increase in body weight, the doses of valproic acid and oxcarbazepine were adjusted. At the age of seven, when this case was reported to us, the regimen consisted of 160 mg valproic acid (9 mg/kg) and 180 mg oxcarbazepine (10 mg/kg) twice daily. Epileptic activity had been well controlled since the combined use of valproic acid and oxcarbazepine. No seizures or other epileptic activity were noticed, and the electroencephalogram at the age of seven showed only scarce sharp waves during sleep (Figure 2).
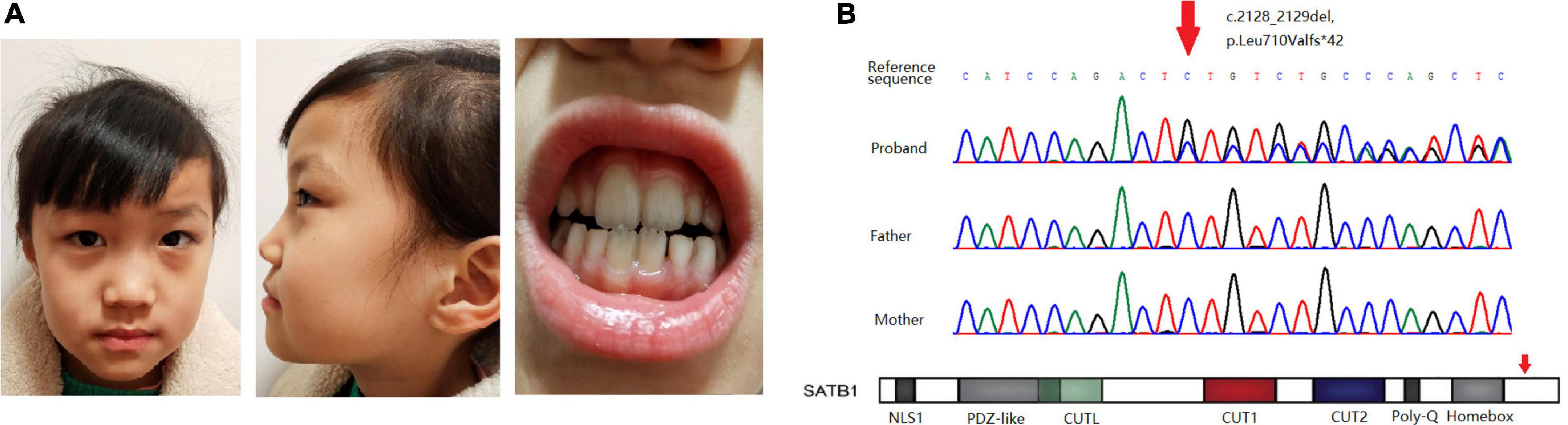
Figure 1. (A) Facial features and dental abnormalities are not recognizable in the patient. (B) Sanger sequencing of the variant in the pedigree and location of the variant in the gene.
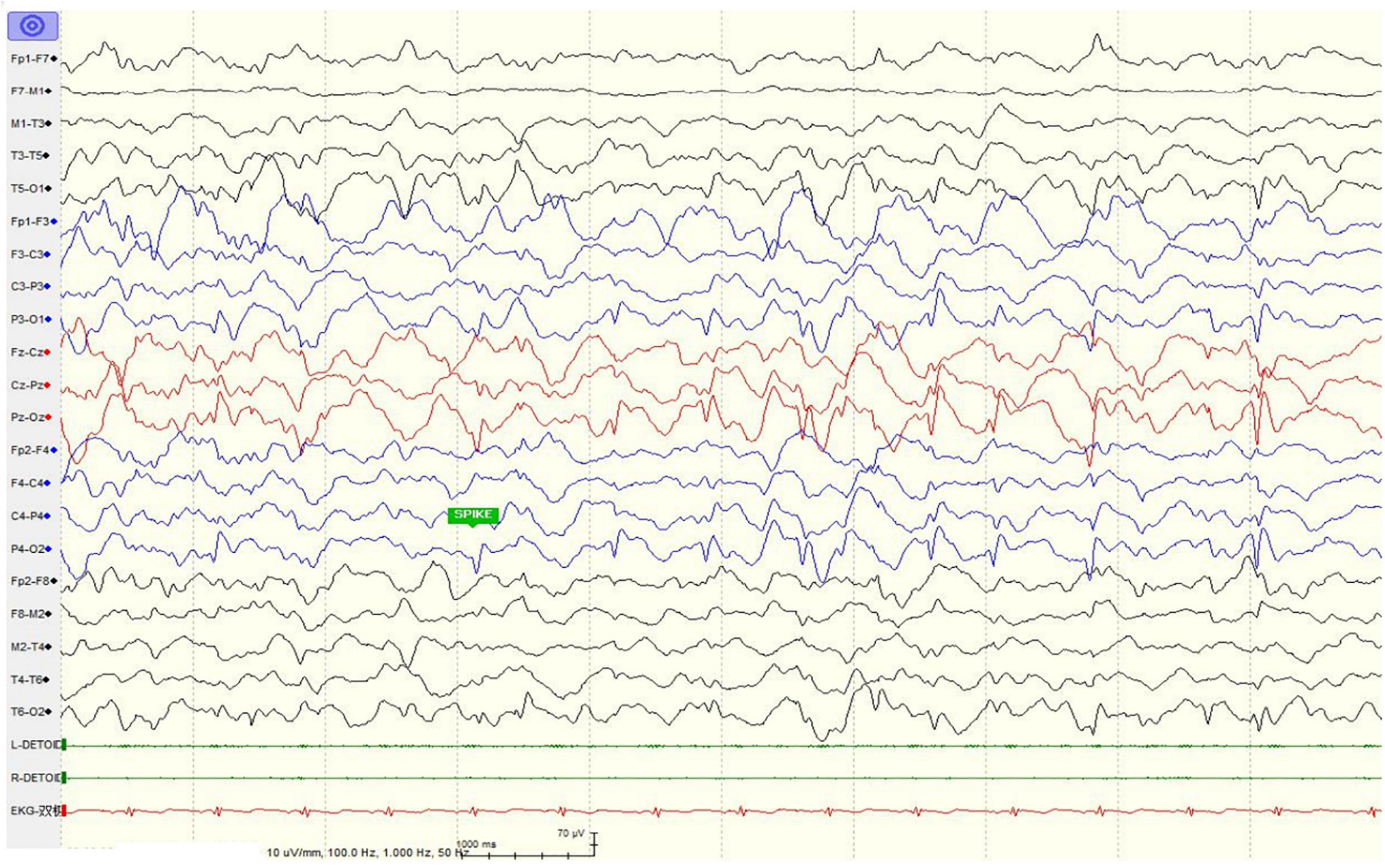
Figure 2. Electroencephalogram of the patient showed only scarce sharp waves during sleep at age seven after anti-epileptic drug treatment.
The Wechsler Intelligence Scale for Children, Fourth Edition (WISC-IV) was used to evaluate the general intellectual ability of the patient when she was 78 months old. The full-scale intelligence quotient was 78, which is at the lower limit of the normal range. The scores of verbal comprehension, perceptual reasoning, working memory, and processing speed subtests were 81, 79, 85, and 79, respectively. Detailed neurological features are shown in Table 1.
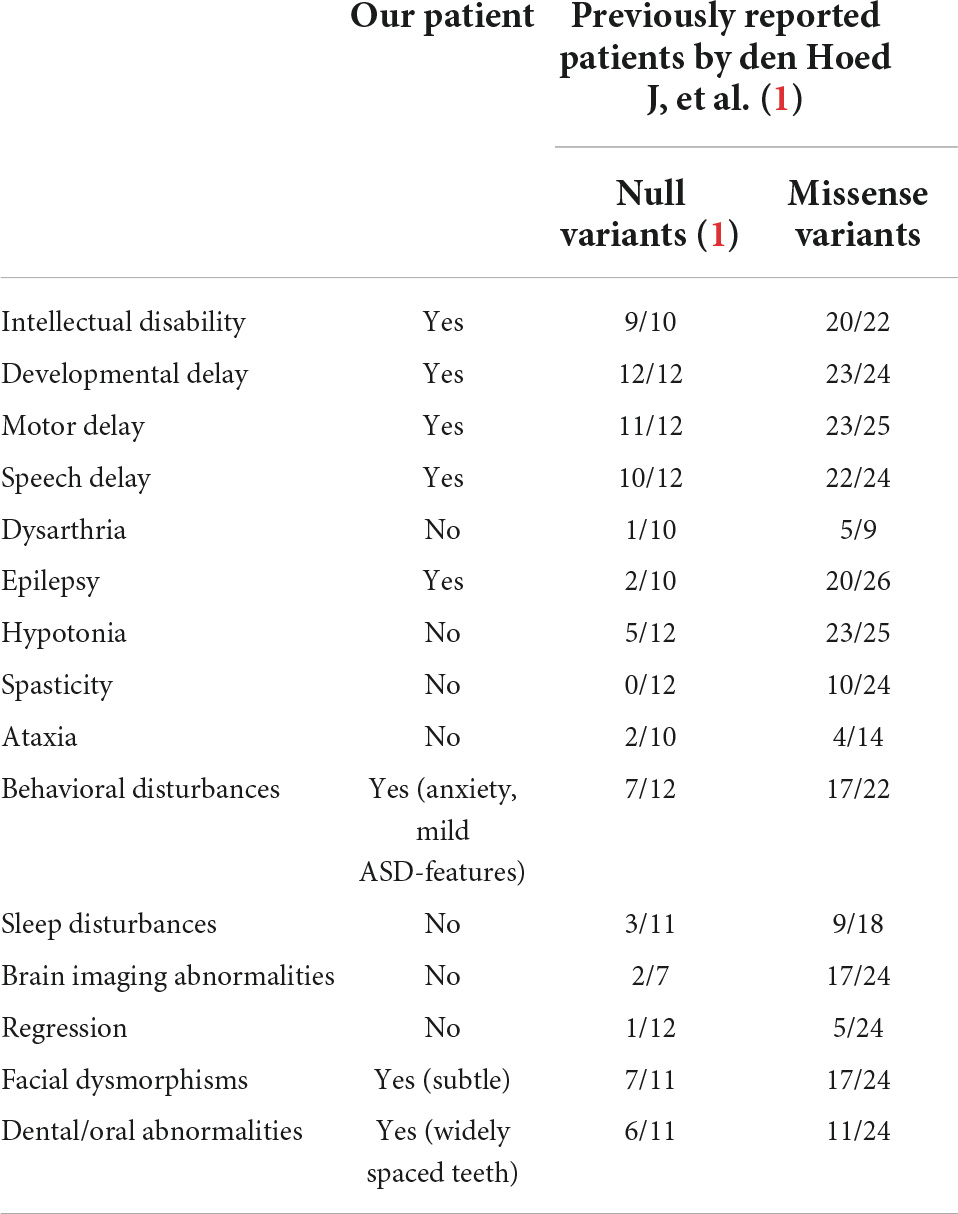
Table 1. Neurological clinical features of patients with SATB1 gene variants.
To detect disease-causing mutations, genomic DNA was extracted from the peripheral blood samples of the patient and her parents using the Gentra Puregene Blood Kit (Qiagen, Hilden, Germany), according to the manufacturer’s protocol. Whole-exome capture was performed using an Agilent SureSelect V6 enrichment capture kit (Agilent Technologies, Inc., Woburn, MA, United States), according to the manufacturer’s instructions. The captured library was sequenced using the Illumina HiSeq 2500 System (Illumina, Inc., San Diego, CA, United States). Original sequencing data were assessed using FastQC (version 0.11.2) for quality control. The Burrows Wheeler alignment tool v0.2.10 was used for sequencing data alignment to the Human Reference Genome (NCBI build 37, hg 19). Single-nucleotide variants and small indels were identified using the Genome Analysis Toolkit. All variants were saved in VCF format and uploaded to the Ingenuity Variant Analysis (Ingenuity Systems, Redwood City, CA, United States) and TGex (Translational Genomics Expert) platforms for biological analysis and interpretation, as previously reported (4). Variants detected by next-generation sequencing were confirmed by Sanger sequencing.
Whole-exome sequencing revealed a novel frame-shift variant, c.2128_2129del, p.Leu710Valfs × 42, in the last exon of SATB1 (NM_001195470.2). Sanger sequencing confirmed the variant as well as the wild-type status of her father and mother (Figure 1B). The control population database (gnomAD) and our local control cohort database did not have reports on the detected variant. The frame-shift variant is classified as a pathogenic variant according to the ACMG guidelines for variant interpretation.
SATB1 (OMIM × 602075) encodes a transcription factor involved in T cell development and maturation (5). The pathogenicity of SATB1 variants was initially reported upon the identification of de novo variants in two large neurodevelopmental disorder cohorts. These variants suggested a role for this gene in neurodevelopment (6, 7). Accurate genotype-phenotype correlations and disease mechanisms were recently identified during the clinical evaluation of a 42-patient cohort (1). Although the broad phenotypic spectrum of SATB1 mutations has been described (including neurodevelopmental delay, intellectual disability, muscle tone abnormalities, epilepsy, behavioral problems, facial dysmorphisms, and dental abnormalities), the treatment and prognosis of these patients have not been reported previously.
Epilepsy is the only disease symptom for which systematic drug regimens are available, as more than 25 antiseizure medications are currently used. The association between epilepsy and neurodevelopmental disorders is well established. Children, especially newborns, infants, and young children, with developmental epilepsies have an increased risk of cognitive, neurobehavioral, and psychiatric disorders (8–10). This has important implications for treatment, especially in children with epileptic encephalopathy, in whom early and successful treatment of seizures and interictal epileptiform activity may be crucial for positive neurodevelopmental outcomes (11, 12). In children with SCN1A seizure disorders, who are at a high risk of sudden unexplained death in epilepsy, seizure control is critical. In addition, prolonged acute seizures may cause permanent injury. In Dravet syndrome, cognitive deterioration may occur, especially when seizure control is incomplete (3, 13). A beneficial effect of immunotherapy combined with anti-epileptic drugs on seizure frequency and cognition has been observed in patients (14). A strong link between seizure control and improvement in neurological function has been observed in many common and rare epilepsy syndromes (15, 16). The combination of low-dose valproic acid and oxcarbazepine showed satisfactory effects on the control of epilepsy in our patient; her neurodevelopment also improved since she was seizure-free. Satb1 was expressed in midbrain dopaminergic neurons and acted as a dopaminergic neuron-specific regulator (17, 18). The attenuation effect of dopaminergic neurotoxicity by valproic acid was assumed to be effective (19). Oxcarbazepine might effectively reduce seizure frequency when used as an add-on for drug-resistant epilepsy (20), but the mechanism by which valproic acid and oxcarbazepine combination works in patients with SATB1 variants needs further investigation. Treatment process and prognosis of our case could provide reliable reference for management of patients with same gene mutation.
A clear genotype-phenotype correlation has been observed: individuals carrying missense variants were more severely affected than individuals carrying protein-truncating variants (1). Functional assays using cells expressing pathogenic variants of SATB1 harboring missense mutations in the CUT1 and CUT2 DNA-binding domains demonstrated altered transcriptional activity compared with the wild-type protein, which could explain this genotype-phenotype correlation (1). Although the de novo frame-shift variant detected in our patients was located in the last exon of the gene, it is likely to escape nonsense-mediated mRNA decay, as a variant located at a more distal position has been reported (p.N736fs × 8). Our patient showed a mild phenotype consistent with previously reported patients presenting protein-truncating variants as a distinct group given the haploinsufficiency of these mutations (Table 1). Diagnosis in patients with mild to moderate developmental delay without evident facial dysmorphism and dental/oral abnormalities is difficult; thus, genetic testing in these patients is quite effective for diagnostic purposes.
We report a de novo protein-truncating variant of SATB1 in a Chinese patient with epilepsy, developmental delay, and dysmorphic features. The clinical features of our patient are consistent with previously reported genotype-phenotype correlations. The anti-epileptic drug treatment of the patient is described in detail, providing important information for the control of epilepsy in patients with SATB1 variations, which is crucial for their neurodevelopment.
The data presented in this study are deposited in the GSA for human repository, accession number SubHRA003891.
The studies involving human participants were reviewed and approved by Sanya Women and Children’s Hospital Managed by Shanghai Children’s Medical Center. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
YY, CL, JeW, and RY contributed to the conception and design of the study. CL and WL organized the database. YY and DW performed the statistical analysis. RY and YY wrote the first draft of the manuscript. CL, JaW, and LC wrote sections of the manuscript. All authors contributed to the manuscript revision and read and approved the submitted version.
This project was supported by grant from the Health Commission of Hainan Province, China (21A200330), grant from Hainan Province Clinical Medical Center, and the Golden Coconut Seed Funding (JYZZ-YJ-202113).
We would like to give the patient’s family great thanks.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. den Hoed J, de Boer E, Voisin N, Dingemans AJM, Guex N, Wiel L, et al. Mutation-specific pathophysiological mechanisms define different neurodevelopmental disorders associated with SATB1 dysfunction. Am. J. Hum. Genet. (2021) 108:346–56. doi: 10.1016/j.ajhg.2021.01.007
2. Picard F, Makrythanasis P, Navarro V, Ishida S, de Bellescize J, Ville D, et al. DEPDC5 mutations in families presenting as autosomal dominant nocturnal frontal lobe epilepsy. Neurology. (2014) 82:2101–6. doi: 10.1212/WNL.0000000000000488
3. Takayanagi M, Haginoya K, Umehara N, Kitamura T, Numata Y, Wakusawa K, et al. Acute encephalopathy with a truncation mutation in the SCN1A gene: a case report. Epilepsia. (2010) 51:1886–8. doi: 10.1111/j.1528-1167.2010.02600.x
4. Yao R, Yu T, Xu Y, Yu L, Wang J, Wang X, et al. Clinical presentation and novel pathogenic variants among 68 Chinese neurofibromatosis 1 children. Genes. (2019) 10:847.
5. Ahlfors H, Limaye A, Elo LL, Tuomela S, Burute M, Gottimukkala KVP, et al. SATB1 dictates expression of multiple genes including IL-5 involved in human T helper cell differentiation. Blood. (2010) 116:1443–53. doi: 10.1182/blood-2009-11-252205
6. Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. (2020) 180:568–84. doi: 10.1016/j.cell.2019.12.036
7. Kaplanis J, Samocha KE, Wiel L, Zhang Z, Arvai KJ, Eberhardt RY, et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. (2020) 586:757–62. doi: 10.1038/s41586-020-2832-5
8. Seidenberg M, Pulsipher DT, Hermann B. Cognitive progression in epilepsy. Neuropsychol Rev. (2007) 17:445–54.
9. Hermann B, Seidenberg M, Jones J. The neurobehavioural comorbidities of epilepsy: can a natural history be developed? Lancet Neurol. (2008) 7:151–60. doi: 10.1016/S1474-4422(08)70018-8
10. Plioplys S, Dunn DW, Caplan R. 10-year research update review: psychiatric problems in children with epilepsy. J Am Acad Child Adolesc Psychiatry. (2007) 46:1389–402. doi: 10.1097/chi.0b013e31815597fc
11. Bombardieri R, Pinci M, Moavero R, Cerminara C, Curatolo P. Early control of seizures improves long-term outcome in children with tuberous sclerosis complex. Eur J Paediatr Neurol. (2010) 14:146–9.
12. Barone V, van Putten MJAM, Visser GH. Absence epilepsy: Characteristics, pathophysiology, attention impairments, and the related risk of accidents. A narrative review. Epilepsy Behav. (2020) 112:107342. doi: 10.1016/j.yebeh.2020.107342
13. Hipaux M, Villeneuve N, Sabouraud P, Desguerre I, Boddaert N, Depienne C, et al. Unusual consequences of status epilepticus in Dravet syndrome. Seizure. (2010) 19:190–4. doi: 10.1016/j.seizure.2010.01.007
14. Hansen N, Widman G, Witt JA, Wagner J, Becker AJ, Elger CE, et al. Seizure control and cognitive improvement via immunotherapy in late onset epilepsy patients with paraneoplastic versus GAD65 autoantibody-associated limbic encephalitis. Epilepsy Behav. (2016) 65:18–24. doi: 10.1016/j.yebeh.2016.10.016
15. Dirani M, Nasreddine W, Abdulla F, Beydoun A. Seizure control and improvement of neurological dysfunction in Lafora disease with perampanel. Epilepsy Behav Case Rep. (2014) 2:164–6. doi: 10.1016/j.ebcr.2014.09.003
16. Curatolo P, Nabbout R, Lagae L, Aronica E, Ferreira JC, Feucht M, et al. Management of epilepsy associated with tuberous sclerosis complex: updated clinical recommendations. Eur J Paediatr Neurol. (2018) 22:738–48.
17. Huang Y, Zhang L, Song NN, Hu ZL, Chen JY, Ding YQ. Distribution of Satb1 in the central nervous system of adult mice. Neurosci Res. (2011) 71:12–21. doi: 10.1016/j.neures.2011.05.015
18. Cancio-Bello A, Saez-Atienzar S. SATB1 is a dopaminergic neuron-specific regulator of cellular senescence. Mov Disord. (2020) 35:235. doi: 10.1002/mds.27978
19. Chen PS, Wang CC, Bortner CD, Peng GS, Wu X, Pang H, et al. Valproic acid and other histone deacetylase inhibitors induce microglial apoptosis and attenuate lipopolysaccharide-induced dopaminergic neurotoxicity. Neuroscience. (2007) 149:203–12. doi: 10.1016/j.neuroscience.2007.06.053
Keywords: SATB1, epilepsy, anti-epileptic drugs, neurodevelopmental delay, protein-truncating variants
Citation: Yu Y, Li C, Li W, Chen L, Wang D, Wang J, Wang J and Yao R (2022) Neurodevelopmental disorders and anti-epileptic treatment in a patient with a SATB1 mutation: A case report. Front. Pediatr. 10:931667. doi: 10.3389/fped.2022.931667
Received: 29 April 2022; Accepted: 08 August 2022;
Published: 02 September 2022.
Edited by:
Hong Ni, Children’s Hospital of Soochow University, ChinaReviewed by:
Jianxiang Liao, Shenzhen Children’s Hospital, ChinaCopyright © 2022 Yu, Li, Li, Chen, Wang, Wang, Wang and Yao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Wang, bGFid2FuZ2ppYW5Ac2hzbXUuZWR1LmNu; Ruen Yao, eWFvcnVlbkAxMjYuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.