 Zhechi He
Zhechi He Ke Wu
Ke Wu Wenqing Xie1
Wenqing Xie1 Jianghua Chen
Jianghua Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 25 August 2022
Sec. Genetics of Common and Rare Diseases
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.930258
Background: Focal segmental glomerulosclerosis (FSGS) is a histopathological diagnosis of the sclerosis of glomeruli and the damage to renal podocytes. FSGS affects the filtration function of the kidneys and results in nephrotic syndrome (NS) in children and adults. FSGS is a clinically and genetically heterogeneous disorder. FSGS-1 [OMIM #603278] is one of the progressive hereditary renal diseases. It is caused by heterozygous variants of the actinin alpha 4 (ACTN4) [OMIM*604638] gene on chromosome 19q13.2 in a dominant inheritance (AD) manner. With the recent development of whole-exome sequencing (WES), 22 (including our case) pathogenic or likely pathogenic variants have been identified in ACTN4 gene.
Case presentation: We reported a 17-year-old Chinese girl who was hospitalized with foamy urine, nausea and vomiting. Laboratory tests revealed increased levels of serum creatinine and urea nitrogen. Ultrasonography demonstrated bilaterally reduced size of kidneys. The primary diagnoses were NS and chronic kidney disease stage 5 (CKD5). The hemodialysis was initiated in 48 h after admission. After 4 months of treatment, the patient received an allogeneic kidney transplantation from her father. A novel heterozygous missense variant c.494C > T (p.A165V) in the ACTN4 gene was found by WES in the patient. This variant was confirmed by Sanger sequencing. The computational simulation of the stability of mutant protein (p.A165V) was decreased. Interatomic interactions of the p.A165V site were increased, and it might be associated with the increased ubiquitylation in the vicinity of the mutant site.
Conclusion: As per the guidelines of the American College of Medical Genetics and Genomics for interpreting sequence variants, the novel heterozygous missense variant was pathogenic (PS2 + PM1 + PM2 + PP3 + PP4). It should be noted that the early onset of severe proteinuria with a poor prognosis is an important and universal symptom for most genetic FSGS. If necessary, genetic screening is recommended.
Focal segmental glomerulosclerosis (FSGS) is a pathological feature of glomerular diseases that presents clinically with proteinuria and a progressive decline in renal function. Patients with FSGS may have initial symptoms of nephrotic syndrome (NS), such as massive proteinuria, hypoalbuminemia, hyperlipidemia, hypertension, and sepsis. FSGS1–FSGS10 are genetically heterogeneous disorders. They are caused by variants in ACTN4 (AD FSGS1 [OMIM #603278]), TRPC6 (AD FSGS2 [OMIM #603965]), CD2AP (susceptibility to FSGS3 [OMIM #607832]), APOL1 (susceptibility to FSGS4 [OMIM #612551]), INF2 (susceptibility to FSGS5 [OMIM #613237]), MYO1E (AR FSGS6 [OMIM #614131]), PAX2 (AD FSGS7 [OMIM #616002]), ANLN (AD FSGS8 [OMIM #616032]), CRB2 (AD FSGS9 [OMIM #616220]), and LMX1B (AD FSGS10 [OMIM #256020]), respectively. Pathogenic variants in the ACTN4 gene are rarely detected in individuals with NS or severe proteinuria. Sadowski et al. (1) showed that disease-causing ACTN4 variants were not found in 2016 individuals with steroid-resistant nephrotic syndrome (SRNS). In addition, Wang et al. (2) also did not detect ACTN4 variants in 110 Chinese children with SRNS. Nagano et al. (3) found that only 2 of 230 Japanese patients with severe proteinuria had ACTN4 variants. Following a review of literature, only 21 pathogenic or likely pathogenic ACTN4 variants were identified. Most variants within the actin binding domain (ABD) of ACTN4 protein contributed to cytoskeleton and podocytes via increasing the binding affinity of ACTN4 to filamentous-actin (F-actin) (4).
In this study, we reported a Chinese girl with a novel pathogenic missense ACTN4 variant affected with end-stage renal disease (ESRD). We conducted a systematic literature review to summarize previously reported clinical phenotypes and ACTN4 variants. Most cases are affected by severe proteinuria at early onset, hypertension, and podocytopathy features.
A 17-year-old girl self-reported nocturia that progressed for half a year. Before being hospitalized, she had noticed foamy urine for 3 weeks. She was admitted following 5 days of nausea and vomiting. Blood tests revealed increased serum creatinine of 18.02 mg/dl, high urea nitrogen, low hemoglobin of 75 g/L, low serum albumin of 28.2 g/L, and severely increased parathyroid hormone (PTH) of 606.0 pg/ml. A lipid panel blood test revealed that the levels of triglyceride (TG), total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), and low-density lipoprotein cholesterol (LDL-C) were normal, and the level of very-low-density lipoprotein (VLDL) was decreased a bit. A urine analysis showed an increased level of proteinuria of 10.85/L/day, a positive result of a hematuria blood test, and a high level of pro-B-type natriuretic peptide (>9,000 pg/ml). In the meantime, there was no significant change in her weight. She did not has mental development delay. Ultrasonography revealed the reduced size of bilateral kidneys with hyperechoic reflections inside the renal parenchyma. A kidney biopsy was offered, but it was refused by her parents. Echocardiography indicated small accumulations of pericardial fluid, the mildly regurgitant tricuspid valve, and a slightly enlarged left ventricle. The primary diagnoses were NS, CKD5, renal anemia, and secondary hyperparathyroidism. After admission, the hemodialysis was initiated in 48 h and continued for 4 months. Then, the patient received an allogeneic kidney transplant from her father.
The patient was the third child of phenotypically normal Chinese parents. The other two siblings were healthy and proteinuria was not found (Figure 1). Her parents did not have consanguineous relation and family history was not remarkable. She was naturally conceived with an uneventful perinatal period. She was born at term at 40 weeks of gestational age. Based on the fact that disease-causing gene variants have been identified in adolescents with idiopathic NS, genetic screening in the patient’s family for inherited diseases was recommended.
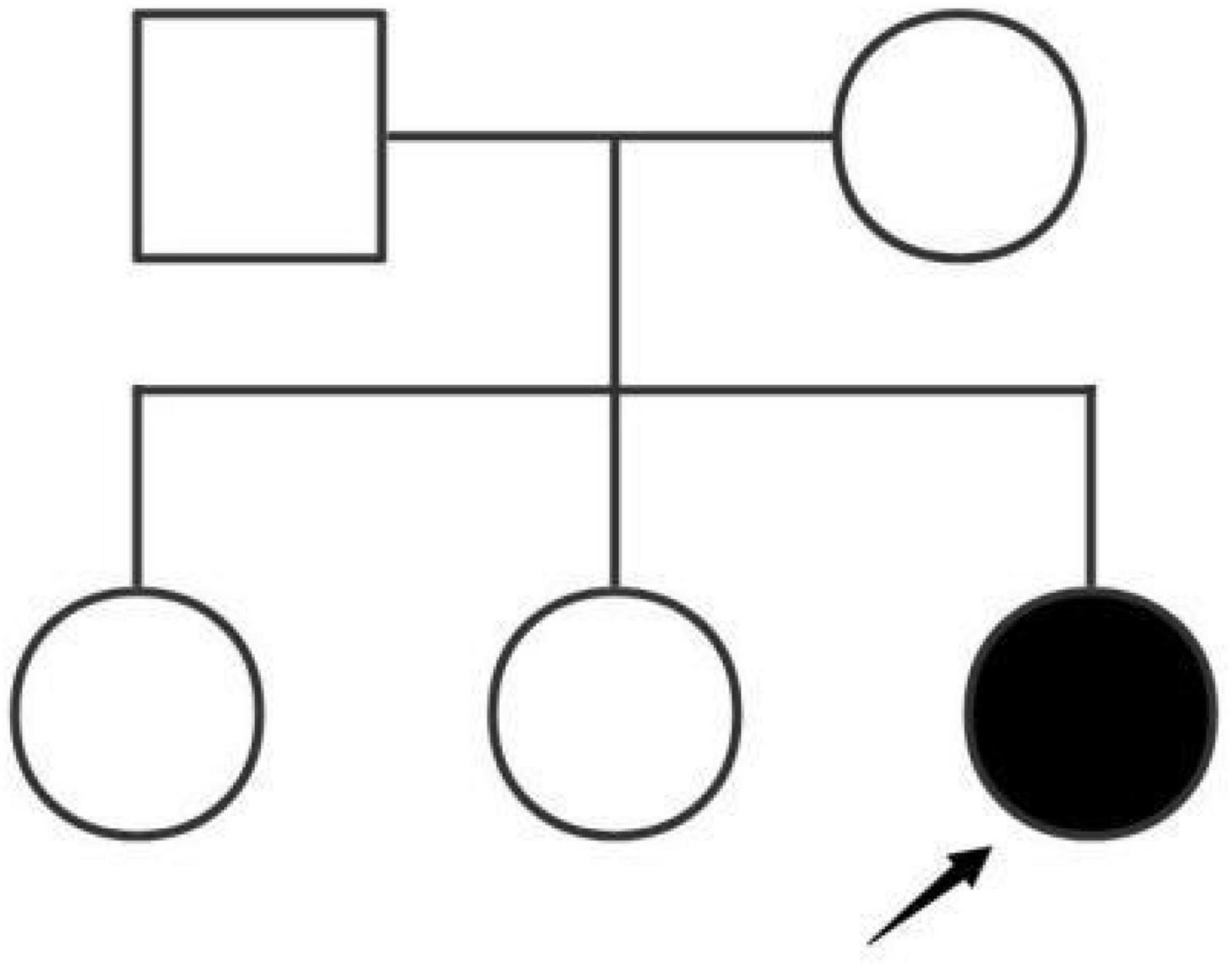
Figure 1. The Chinese patient’s pedigree.
The patient and her parents signed informed consent for genetic analysis. Our legal ethics committee approved this genetic study. Genomic DNA was extracted from the peripheral blood of the patient and phenotypically normal parents for WES. Sanger sequencing was used for further verification. We identified a de novo heterozygous missense variant c.494C > T in the ACTN4 gene (NM_004924.6). This variant has not been registered in population databases (1,000 Genomes Project, gnomAD, and dbSNP) or reported in disease databases (ClinVar, Human Gene Mutation Database, OMIM).
A cosegregation analysis was performed among family members. This variant was de novo and verified by Sanger sequencing (Figure 2). Silico predictive algorithms (SIFT, PolyPhen-2, REVEL, MutPred, MutationTaster, PROVEAN, and MVP) of pathogenicity all showed damage. The analysis of conserved sequences suggested this variant was located in highly conserved sequences across several mammalian species (Figure 3). The variant c.494C > T (p.A165V) was located in a critical and well-established functional domain (the calponin-homology domain of ACTN4 protein) without benign variants (Figure 4). Summing up the above, according to the guidelines of the American College of Medical Genetics and Genomics (ACMG) for interpreting sequence variants, the novel missense variant was pathogenic (PS2 + PM1 + PM2 + PP3 + PP4).
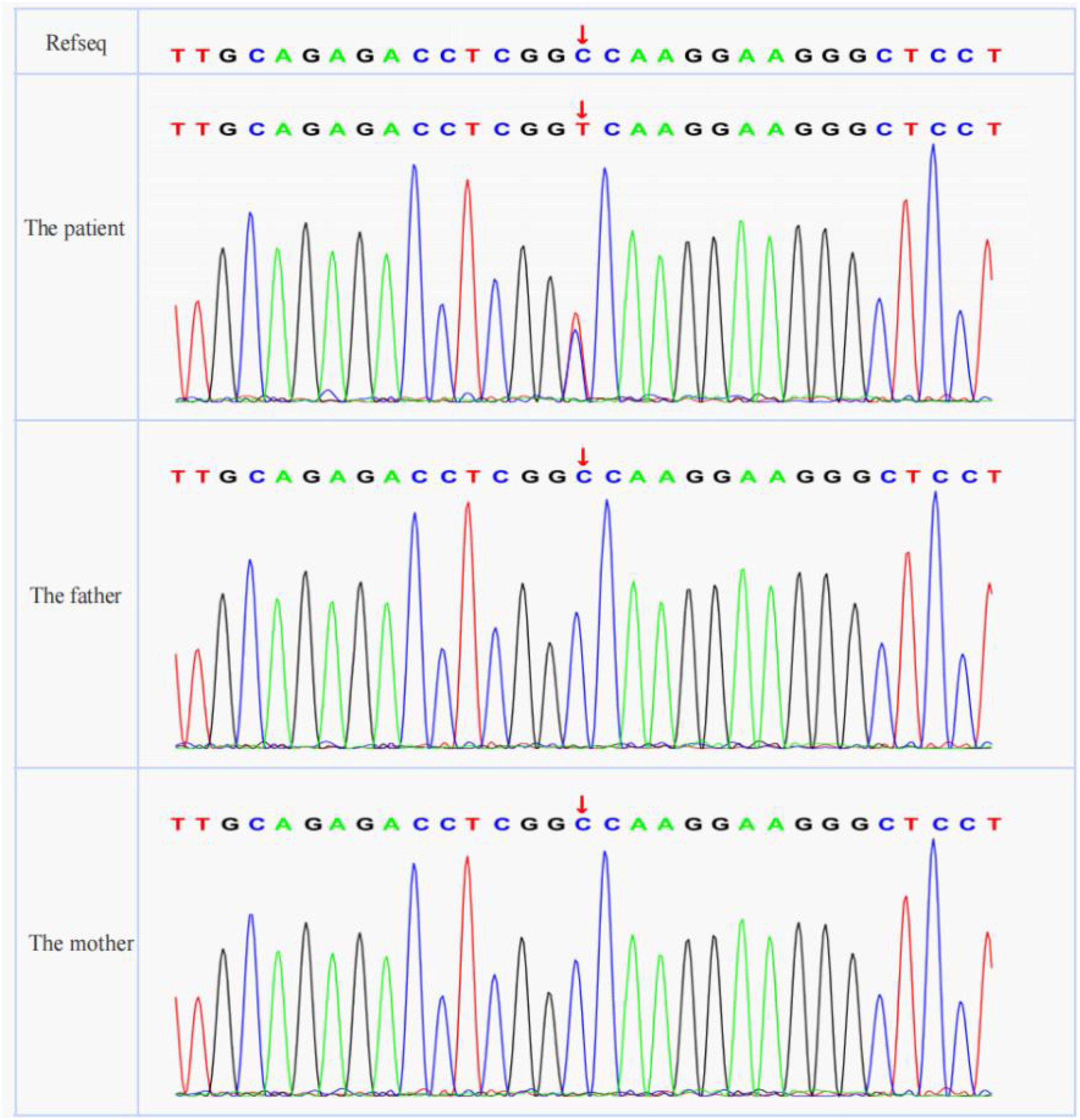
Figure 2. The results of Sanger sequencing (ACTN4 variant was marked with red arrows).
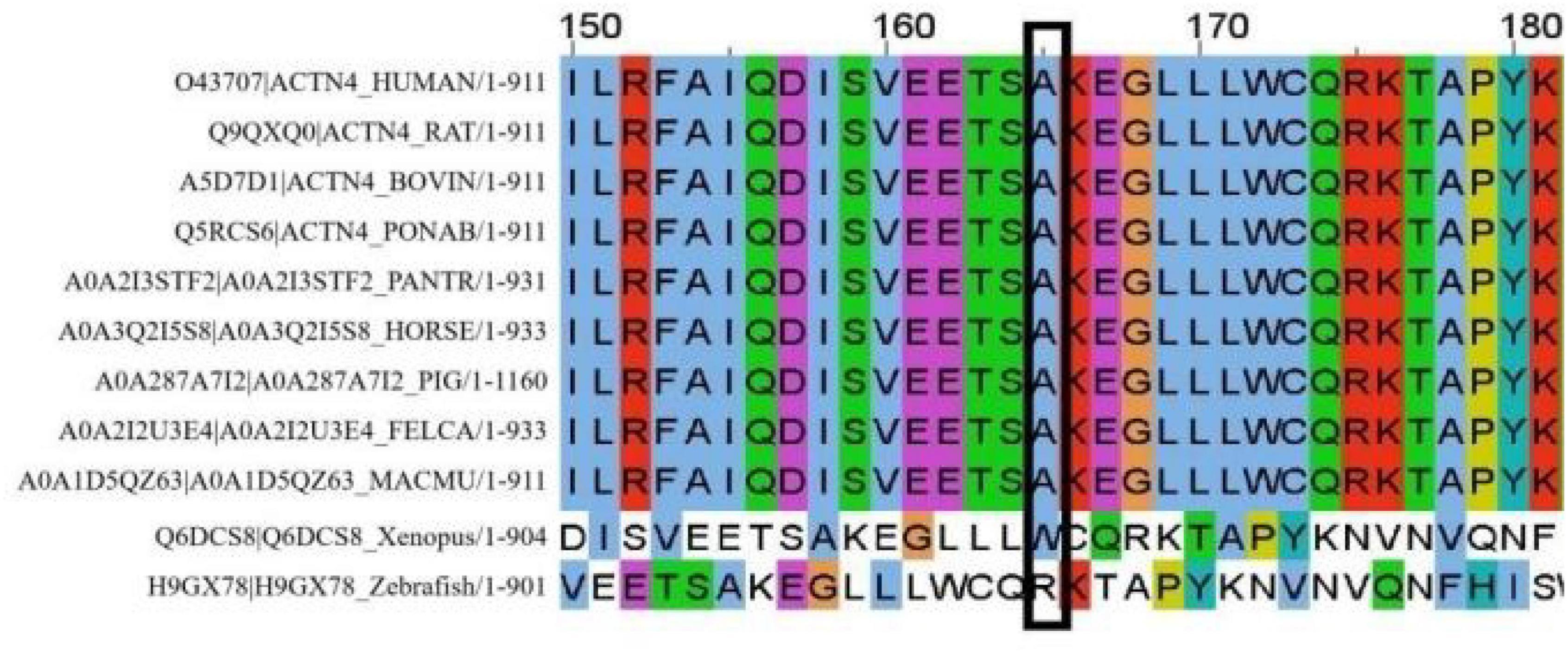
Figure 3. In total, 30 amino acids surrounding the variant position (marked with a black box).
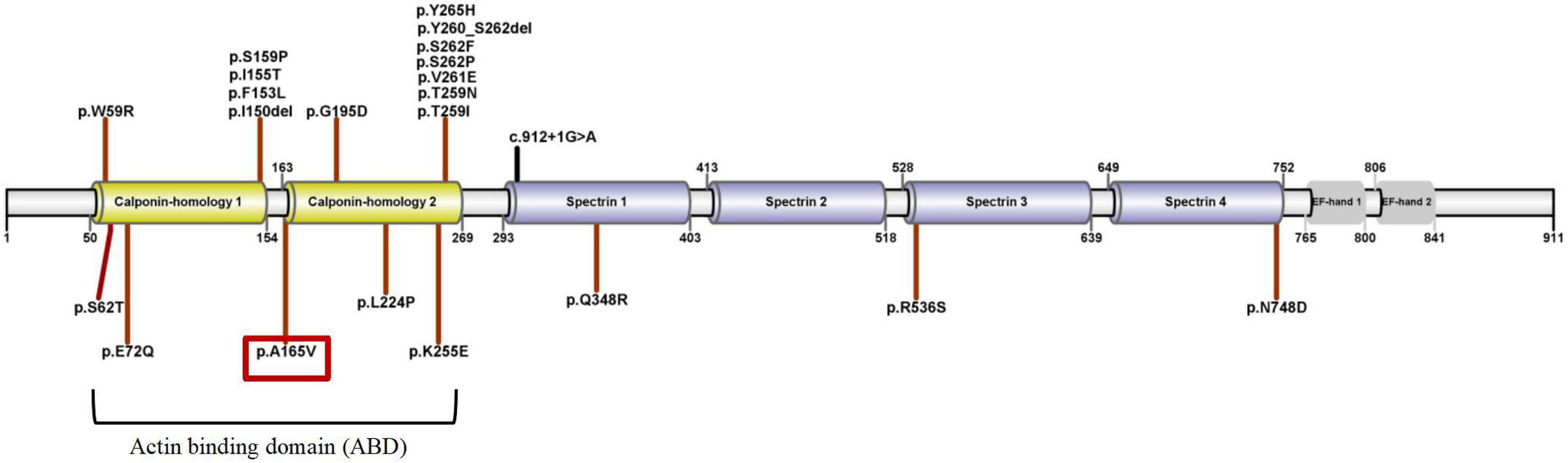
Figure 4. The schematic diagram of ACTN4 variants and ACTN4 protein’s domains (UniProtKB-O43707). The patient’s ACTN4 variant was marked with a red box.
DynaMut (a web server) (5) is a well-established normal mode approach. We used it to visualize and assess the stability and interatomic interactions of mutant proteins. We put information into DynaMut as follows: wild-type structure (PDB accession code: 6O31), mutation detail (A165V and chain A). The prediction outcome of stability was ΔΔG: −0.087 kcal/mol (destabilizing). In comparison with p.A165V, we put p.G195D into DynaMut. The prediction outcome of stability of p.G195D was ΔΔG: −1.349 kcal/mol (destabilizing). The residues 165 and 195 in the wild-type were observed to form residue interactions (colored in light green) with their surrounding residues, whereas some interactions were seen to be increased in mutant sites (p.A165V and p.G195D) (Figure 5).
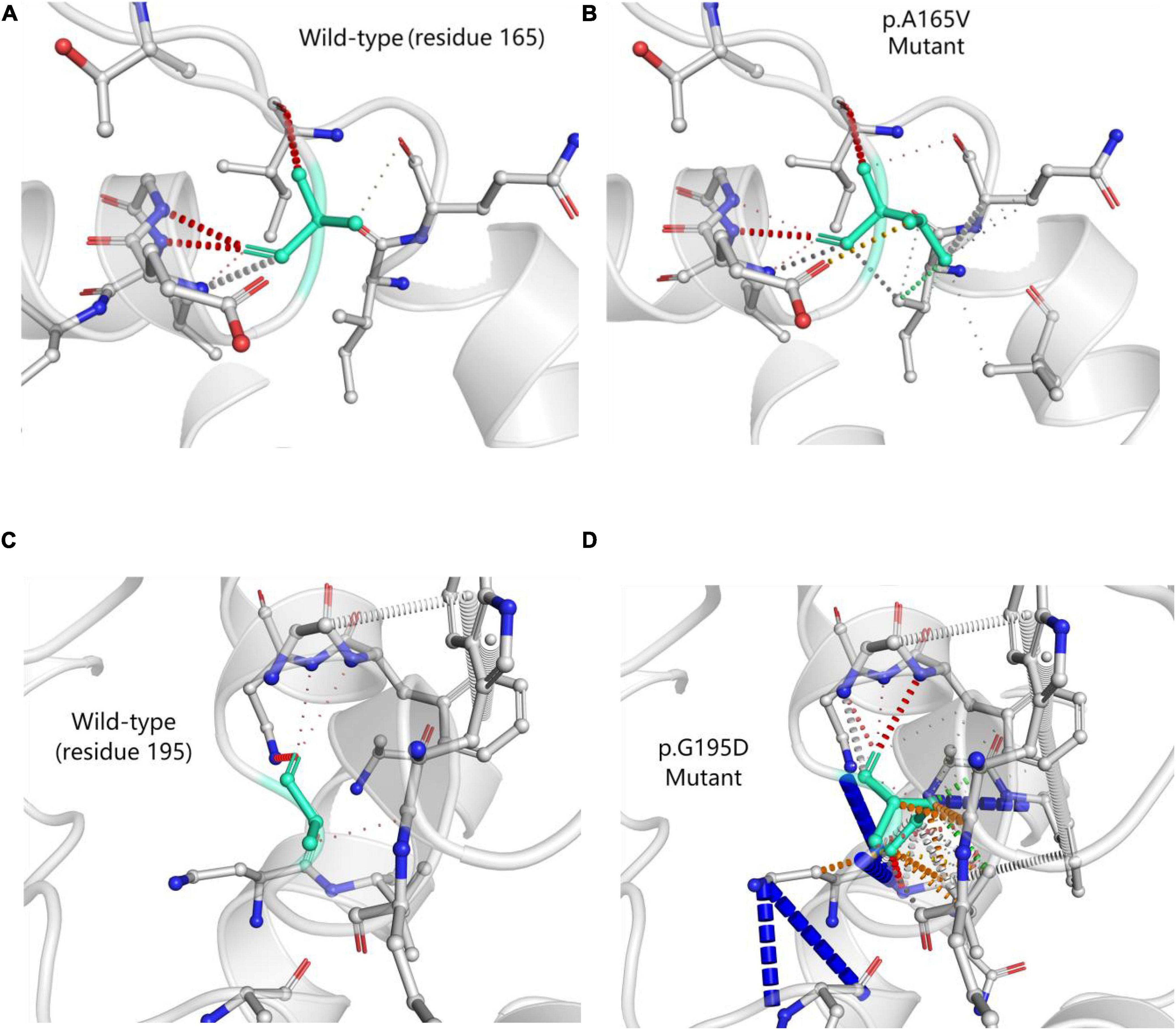
Figure 5. Prediction of interatomic interactions of p.A165V (A,B) and p.G195D (C,D). Residues 165 and 195 in the wild-type and mutant proteins (p.A165V and p.G195D) are colored in light green and are shown as sticks. The respective chemical interactions are labeled as dotted lines and colored as follows: hydrogen bonds—(red), weak hydrogen bonds—(orange), hydrophobic contacts—(green), amide-amide contacts—(blue), and ionic interactions—(gold). Amino acid residues are also colored according to type, namely; nitrogen (blue), oxygen (red), and sulfur (yellow). In comparison with wild-type sites, some interactions were observed to be added in mutant sites.
We searched the PubMed database, the Human Gene Variant Database (HGMD), and Online Mendelian Inheritance in Man (OMIM) using “focal segmental glomerulosclerosis” and “ACTN4” as keywords. The search time was from the establishment of the databases to April 1, 2022. Previous studies with ACTN4 variants and clinical characteristics were reviewed. In total, fifteen documents were retrieved. Although 41 mutations were registered in HGMD, we reclassified the pathogenicity of these variants according to standards and guidelines for the interpretation of sequence variants. A total of 22 pathogenic or likely pathogenic ACTN4 variants and related phenotypes are summarized in Table 1 and Figure 4. Three heterozygous variants [c.1-34C > T, c.1-590delA, and (1-1044delT) + (1-797T > C) + (1-769A > G)] in the promoter of the ACTN4 gene have been reported in patients with FSGS. The functional analysis of these promoter variants indicated that they might also contribute to the pathophysiology of idiopathic FSGS (6). The remaining 17 known variants are shown in Table 2. More importantly, variants in the ACTN4 gene were missense and occurred de novo. Most patients with ACTN4 variants had proteinuria at adolescent onset and rapidly progressed to ESRD.
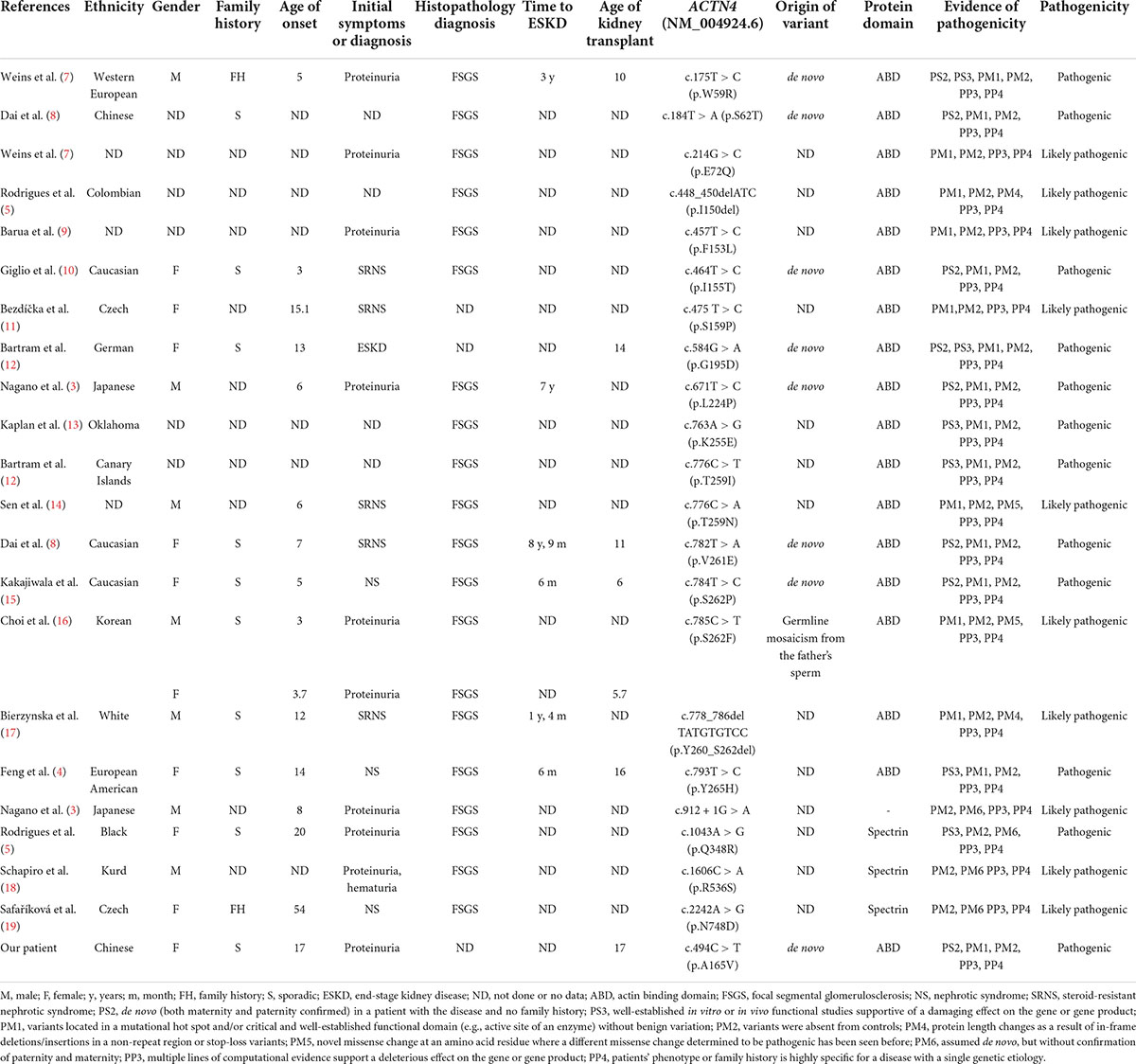
Table 1. Genotypes and phenotypes of patients with ACTN4 variants previously reported.

Table 2. The known variants of VUS in the ACTN4 gene.
The human ACTN4 gene is located on chromosome 19q13.2. The transcript of ACTN4 (NM_004924.6) has 21 exons, a transcript length of 4,990 base pairs, and a translation length of 911 amino acids. The alpha-actinin-4 protein (ACTN4) [UniProtKB-O43707] is one member of four actin-binding proteins. ACTN4 is ubiquitously expressed in different tissues and highly expressed in the placenta and kidney (20). ACTN4 was prominently distributed in podocytes via immunostaining followed by confocal microscopy (21).
Actinin alpha 4 has three main domains: (1) the N-terminal highly conserved ABD consists of two calponin homology (CH1/2); (2) spectrin 1-4; and (3) the C-terminal domain consists of EF-hand 1/2 (Figure 4).
Most of the pathogenic or likely pathogenic variants were missense and located within ABD. ACTN4 variants in ABD damaged the podocyte foot process architecture and function through a “gain-of-function” mechanism (increased the binding of ACTN4 to F-actin). It was a “dominant effect” that mutant ACTN4 protein unconventionally aggregated in the cell and the binding affinity of ACTN4 to F-actin was increased (22). Bartram et al. (12) identified p.G195D in a patient with juvenile onset of proteinuria and ESRD. A functional analysis of p.G195D showed that ACTN4 was the significantly reduced protein in the patient’s primary renal epithelial cells. Further analysis revealed a “dominant effect” in which the decreased stability of mutant protein was associated with the increased ubiquitylation in the vicinity of p.G195D site. The aggregated unstable mutant protein led to the disturbance of the renal cell cytoskeleton. The p.G195D and p.A165V were all located within CH2 domain of ACTN4. So we hypothesized that p.A165V may have the same pathogenic mechanism as p.G195D. The computational simulation of p.G195D seemed to accord closely with the functional analysis of p.G195D. All results showed that the stability of the mutant proteins (p.G195D and p.G195D) was decreased. The stability of the mutant protein might be mediated by the increased ubiquitylation. Therefore, it was tempting to speculate that the increased ubiquitylation in the vicinity of mutant sites may be connected with increased interatomic interactions of mutant sites. Presumably, p.A165V and p.G195D had a basic similarity in the pathogenic mechanism.
Three variants (Table 1) occurred in spectrin domains that were known to mediate CLP36-binding. Functional studies showed ACTN4 variants in spectrin domains provided a plausible “loss-of-function” mechanism (decreased the binding of ACTN4 to CLP36). Liu et al. (23) reported that deficiencies of ACTN4 and CLP36 were observed in podocytes of FSGS cases by performing immunohistochemical staining. The mutant protein (p.Q348R) significantly inhibited the ability of ACTN4 to interact with CLP36. Depletion of the level of CLP36 or disruption of the ACTN4-CLP36 complex significantly impeded RhoA activity and the generation of traction force in podocytes but did not alter the actin-binding affinity (12).
The C-terminal EF-hand domain is involved in binding intracellular Ca2+. ACTN4 has two isoforms (Ca2+-sensitive and Ca2+-insensitive). The Ca2+-sensitive isoform is broadly expressed, while the Ca2+-insensitive isoform is predominantly expressed in the nervous system (24). The binding of Ca2+ to EF-hand domains reduced the affinity of F-actin binding (25). Five variants in HGMD occurred in EF-hand domains. Safaříková et al. (19) found p.A784V, p.P787L, p.C793Y, and p.G798D in patients with IgA nephropathy. We reclassified these four variants as uncertain significance. Chatterjee et al. (26) reported p.V801M in a patient with proteinuria. It was reclassified as a variant of uncertain significance. For now, no pathogenic or likely pathogenic variants have been identified in the C-terminal EF-hand domains.
To our knowledge, ACTN4 variants in literature have all occurred de novo (except no data were collected). ACTN4 variants within ABD showed a “gain-of-function” mechanism. The homozygous Actn4 knockout (Actn4 KO) mice developed albuminuria and FSGS at about 10 weeks of age, loss of Actn4 damaged glomerular podocytes and filtration barrier (13). A plausible “loss-of-function” mechanism of ACTN4 variants within spectrin or EF-hand domains also existed. The extent of the reduction of ACTN4 may be closely associated with the severity of proteinuria in FSGS cases. As shown in Table 1, patients (14/16) with ACTN4 variants within ABD were affected with proteinuria before the age of 18 years and developed rapidly progression to ESRD. Patients (2/16) with ACTN4 variants within spectrin domains appeared proteinuria over 18 years old (one case was 54 years old). It needs more cases to verify whether or not two different mechanisms are associated with the age of onset.
In total, 17 known ACTN4 variants were of unknown significance (Table 2). As per the guidelines of ACMG for interpreting sequence variants, although these variants were absent from normal controls, multiple silico predictive algorithms suggested no impact on these variants (conservation, evolutionary, splicing impact, etc.) or reputable source recently reported these variants as likely benign or benign variants. The phenotypes of some patients with these variants were not associated with FSGS. This summary (Table 2) might note that phenotypic variability of ACTN4-mediated diseases. Whereas, it is restricted to a small amount of reported cases. So, clinicians should be cautious of dealing with these variants.
The genetic screening is recommended for those juveniles who are accidentally found to have severe proteinuria. Although we are realistic that no treatment is available for ACTN4-mediated FSGS, a genetic screening can help physicians and patients to prepare the kidney transplantation in time. If parents consider to give birth to another child, a definitive genetic diagnosis is essential for prenatal diagnosis.
In conclusion, we described a Chinese patient with a novel pathogenic ACTN4 variant. The genetic screening of congenital proteinuria is necessary for patients. We summarized the pathogenicity of ACTN4 variants in previous literature. The severe proteinuria at early onset and rapid progression to ESRD are universal and important symptoms of ACTN4-mediated FSGS, and those patients are good candidates for the kidney transplantation.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the Ethical Committee of The First Affiliated Hospital, Zhejiang University School of Medicine. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
KW wrote the main manuscript text and critically revised the manuscript. WX carried out the molecular genetic experimental. ZH and JC prepared the clinical data and imaging data. ZH and KW contributed to the check of revision, genetic evaluation, and genetic databases analysis. All authors reviewed the manuscript and read and approved the final manuscript.
The authors declared that they received funding from the Institute of Nephrology, Zhejiang Clinical Research Center of Kidney and Urinary System Disease (2021E50001).
We are grateful to the patient and her family for participation in this study, as well as all the physicians in the course of the medical treatment. We thank the staff of Chigene (Beijing) Translational Medical Research Center Co. Ltd. for assisting with WES and Sanger sequencing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. (2015) 26:1279–89. doi: 10.1681/ASN.2014050489
2. Wang F, Zhang Y, Mao J, Yu Z, Yi Z, Yu L, et al. Spectrum of mutations in Chinese children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. (2017) 32:1181–92. doi: 10.1007/s00467-017-3590-y
3. Nagano C, Yamamura T, Horinouchi T, Aoto Y, Ishiko S, Sakakibara N, et al. Comprehensive genetic diagnosis of Japanese patients with severe proteinuria. Sci Rep. (2020) 10:270. doi: 10.1038/s41598-019-57149-5
4. Feng D, Steinke JM, Krishnan R, Birrane G, Pollak MR. Functional validation of an alpha-actinin-4 mutation as a potential cause of an aggressive presentation of adolescent focal segmental glomerulosclerosis: Implications for genetic testing. PLoS One. (2016) 11:e0167467. doi: 10.1371/journal.pone.0167467
5. Rodrigues CH, Pires DE, Ascher DB. DynaMut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. (2018) 46:W350–5. doi: 10.1093/nar/gky300
6. Dai S, Wang Z, Pan X, Wang W, Chen X, Ren H, et al. Functional analysis of promoter mutations in the ACTN4 and SYNPO genes in focal segmental glomerulosclerosis. Nephrol Dial Transplant. (2010) 25:824–35. doi: 10.1093/ndt/gfp394
7. Weins A, Kenlan P, Herbert S, Le TC, Villegas I, Kaplan BS, et al. Mutational and biological analysis of alpha-actinin-4 in focal segmental glomerulosclerosis. J Am Soc Nephrol. (2005) 16:3694–701. doi: 10.1681/ASN.2005070706
8. Dai S, Wang Z, Pan X, Chen X, Wang W, Ren H, et al. ACTN4 gene mutations and single nucleotide polymorphisms in idiopathic focal segmental glomerulosclerosis. Nephron Clin Pract. (2009) 111:c87–94. doi: 10.1159/000191198
9. Barua M, Brown EJ, Charoonratana VT, Genovese G, Sun H, Pollak MR, et al. Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis. Kidney Int. (2013) 83:316–22. doi: 10.1038/ki.2012.349
10. Giglio S, Provenzano A, Mazzinghi B, Becherucci F, Giunti L, Sansavini G, et al. Heterogeneous genetic alterations in sporadic nephrotic syndrome associate with resistance to immunosuppression. J Am Soc Nephrol. (2015) 26:230–6. doi: 10.1681/ASN.2013111155
11. Bezdíčka M, Štolbová S, Seeman T, Cinek O, Malina M, Šimánková N, et al. Genetic diagnosis of steroid-resistant nephrotic syndrome in a longitudinal collection of Czech and Slovak patients: A high proportion of causative variants in NUP93. Pediatr Nephrol. (2018) 33:1347–63. doi: 10.1007/s00467-018-3950-2
12. Bartram MP, Habbig S, Pahmeyer C, Höhne M, Weber LT, Thiele H, et al. Three-layered proteomic characterization of a novel ACTN4 mutation unravels its pathogenic potential in FSGS. Hum Mol Genet. (2016) 25:1152–64. doi: 10.1093/hmg/ddv638
13. Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. (2000) 24:251–6. doi: 10.1038/73456
14. Sen ES, Dean P, Yarram-Smith L, Bierzynska A, Woodward G, Buxton C, et al. Clinical genetic testing using a custom-designed steroid-resistant nephrotic syndrome gene panel: Analysis and recommendations. J Med Genet. (2017) 54:795–804. doi: 10.1136/jmedgenet-2017-104811
15. Kakajiwala AK, Meyers KE, Bhatti T, Kaplan BS. Rapid progression to end-stage renal disease in a child with a sporadic ACTN4 mutation. Clin Nephrol Case Stud. (2015) 3:14–8. doi: 10.5414/CNCS108616
16. Choi HJ, Lee BH, Cho HY, Moon KC, Ha IS, Nagata M, et al. Familial focal segmental glomerulosclerosis associated with an ACTN4 mutation and paternal germline mosaicism. Am J Kidney Dis. (2008) 51:834–8. doi: 10.1053/j.ajkd.2008.01.018
17. Bierzynska A, McCarthy HJ, Soderquest K, Sen ES, Colby E, Ding WY, et al. Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int. (2017) 91:937–47. doi: 10.1016/j.kint.2016.10.013
18. Schapiro D, Daga A, Lawson JA, Majmundar AJ, Lovric S, Tan W, et al. Panel sequencing distinguishes monogenic forms of nephritis from nephrosis in children. Nephrol Dial Transplant. (2019) 34:474–85. doi: 10.1093/ndt/gfy050
19. Safaříková M, Reiterová J, Safránková H, Stekrová J, Zidková A, Obeidová L, et al. Mutational analysis of ACTN4, encoding α-actinin 4, in patients with focal segmental glomerulosclerosis using HRM method. Folia Biol (Praha). (2013) 59:110–5.
20. Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. (2014) 13:397–406. doi: 10.1074/mcp.M113.035600
21. Khurana S, Chakraborty S, Lam M, Liu Y, Su YT, Zhao X, et al. Familial focal segmental glomerulosclerosis (FSGS)-linked α-actinin 4 (ACTN4) protein mutants lose ability to activate transcription by nuclear hormone receptors. J Biol Chem. (2012) 287:12027–35. doi: 10.1074/jbc.M112.345421
22. Feng D, DuMontier C, Pollak MR. The role of alpha-actinin-4 in human kidney disease. Cell Biosci. (2015) 5:44. doi: 10.1186/s13578-015-0036-8
23. Liu Z, Blattner SM, Tu Y, Tisherman R, Wang JH, Rastaldi MP, et al. Alpha-actinin-4 and CLP36 protein deficiencies contribute to podocyte defects in multiple human glomerulopathies. J Biol Chem. (2011) 286:30795–805. doi: 10.1074/jbc.M111.255984
24. Foley KS, Young PW. An analysis of splicing, actin-binding properties, heterodimerization and molecular interactions of the non-muscle α-actinins. Biochem J. (2013) 452:477–88. doi: 10.1042/BJ20121824
25. Pinotsis N, Zielinska K, Babuta M, Arolas JL, Kostan J, Khan MB, et al. Calcium modulates the domain flexibility and function of an α-actinin similar to the ancestral α-actinin. Proc Natl Acad Sci U S A. (2020) 117:22101–12. doi: 10.1073/pnas.1917269117
26. Chatterjee R, Hoffman M, Cliften P, Seshan S, Liapis H, Jain S, et al. Targeted exome sequencing integrated with clinicopathological information reveals novel and rare mutations in atypical, suspected and unknown cases of Alport syndrome or proteinuria. PLoS One. (2013) 8:e76360. doi: 10.1371/journal.pone.0076360
Keywords: focal segmental glomerulosclerosis, end-stage renal disease, proteinuria, ACTN4, de novo
Citation: He Z, Wu K, Xie W and Chen J (2022) Case report and literature review: A de novo pathogenic missense variant in ACTN4 gene caused rapid progression to end-stage renal disease. Front. Pediatr. 10:930258. doi: 10.3389/fped.2022.930258
Received: 27 April 2022; Accepted: 29 July 2022;
Published: 25 August 2022.
Edited by:
Babak Behnam, National Sanitation Foundation International, United StatesCopyright © 2022 He, Wu, Xie and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianghua Chen, Y2hlbmppYW5naHVhQHpqdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.