 Fanghua Ye
Fanghua Ye Hui Zhang
Hui Zhang Wen Zhang
Wen Zhang Liangchun Yang
Liangchun Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 05 December 2022
Sec. Pediatric Hematology and Hematological Malignancies
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.927894
Purpose: Myeloid sarcoma (MS) is a rare extramedullary mass with myeloid expression, which is easy to be missed and misdiagnosed, especially in the pediatric population. We analyze the clinicopathological characteristics, immunophenotypic, cytogenetic, and molecular studies, therapeutic approaches, and outcomes, to optimize the management of such patients.
Methods: A retrospective, single-center, case series study of eleven children diagnosed with MS by pathology was performed.
Results: The male-to-female ratio was 8:3, and the median age at diagnosis was 7 years. The most commonly involved sites were the skin and orbital region, followed by lymph nodes, central nervous system, and testis. Seven cases (64%) with Class I-MS and four cases (36%) presented as Class II-MS. Immunohistochemically, MPO and CD117 were the most commonly expressed markers, followed by CD33, CD43, CD34, CD68, and lysozyme. Chromosomal abnormalities were detected in 4 patients. Two patients had the presence of deleterious mutations (FLT3, ASXL, KIT, and DHX15) on molecular detection. Ten patients were treated with chemotherapy based on AML regimens. The median follow-up time was 33.5 months in eleven patients. Two patients relapsed, one died, and one lost to follow-up. The 2-year overall survival (OS) rate estimated by Kaplan-Meier curves was 90.9% ± 8.7%, and the event-free survival (EFS) rate was 64.9% ± 16.7%.
Conclusions: MS diagnosis is usually challenging. Adequate tumor biopsy and expanded immunohistochemistry are necessary for the correct diagnosis of MS. Early and regular systemic chemotherapy promises long-term survival.
Myeloid sarcoma (MS) is a distinct subtype of hematologic malignancy, defined by the WHO in 2016 as a mass that appears in an anatomical location outside the bone marrow containing myeloid blast cells, commonly involving the skin, soft tissues, central nervous system (CNS), and the urogenital tract (1). MS first described chloroma because of the greenish color, caused by the presence of myeloperoxidase (MPO). The terms granulocytic sarcoma, extramedullary myeloid cell tumor, or myeloblastoma have also been used (2, 3). There are no definitive data on the incidence of MS in the general population. The incidence of MS in acute myeloid leukemia (AML) patients is 2.5%–9.1%, and some literature has also reported that the rate is as high as 40% in children (4, 5). MS may present de novo, may accompany peripheral blood and marrow involvement may present as a relapse of AML, or may present as the progression of a prior myelodysplastic syndrome (MDS), myeloproliferative neoplasms (MPN), or MDS/MPN (1, 6).
The diagnosis of MS is based on clinical manifestations, imaging, mass biopsy, immunohistochemistry, cytogenetics, and molecular biology (7). There are few study reports on MS and no studies on large series. In most patients with known myeloproliferative disorders, the diagnosis of MS may be relatively simple. However, the diagnosis of isolated MS is a challenge.
Here, we report a comprehensive study of MS, including their clinical and laboratory manifestations, pathological and genetic features, treatments, and outcomes, in a series of 11 children. Literature review and single-center experience are used to improve pediatricians' understanding of the disease.
This study was approved by the ethics committee of Xiangya Hospital Central South University in accordance with the Helsinki Declaration. Eleven children diagnosed with MS by pathology of the tumor mass were admitted from January 1, 2016, to April 1, 2022. Diagnoses were reconfirmed by two pathologists. The data we collected included the age, gender, clinical manifestations, site, history of AML or other neoplastic disorders, laboratory inspection, pathologic characteristics, radiological findings, treatment, and outcome.
WHO classifies them according to their extramedullary manifestations (1): Class I, AML with MS; Class II, isolated MS (normal peripheral blood smear and bone marrow morphology and no history of myeloid neoplasm); Class III, extramedullary relapse of AML, including relapse after bone marrow transplantation; Class IV, acute/transformed stage of MDS/MPNs.
The initial diagnosis was made on core or surgical biopsy specimens. Biopsy samples were consistent with the diagnostic criteria of MS (A. Morphological features: The morphology of MS manifests differently depending on the degree of myeloid differentiation, typically showing different stages of myeloid cell infiltration. B. Immunohistochemistry: it mostly showed MPO, CD43, CD34, CD13, CD33, CD117, CD14, CD11b, CD11c, CD68, CD163, lysozyme, and other myeloid and monocyte antigens positive, generally not expressing lymphocyte markers such as CD3, CD19, CD20) (8). Patients with bone marrow and cerebrospinal fluid (CSF) involvement were analyzed by flow cytometry included markers for CD20, CD117, CD34, CD33, CD10, CD45, HLA-DR, CD19, CD15, CD13, CD64, CD7, CD14, CD3, CD123, CD4, CD5, CD11b, and CD38.
The G-banding technique was used to analyze the karyotype. Fluorescent in situ hybridization (FISH) panel including probes for deletion (5)/-5, deletion (7)/-7, t (8;21), t (15;17), and inversion (16), etc. histone lysine [K]-MethylTransferase 2A (KMT2A) gene formerly known as mixed-lineage leukemia (MLL) gene. Polymerase chain reaction (PCR) was used to detect fusion genes including MLL/AF10, AML1/ETO, PML/RARα, ET6V/RUNX1, BCR/ABL1, etc. Myeloid malignancies mutation panel detected in MS tumor mass or marrow by next-generation sequencing (NGS), such as fms like tyrosine kinase 3 (FLT3), tyrosine kinase receptor protein family KIT gene, nucleophosmin 1 (NPM1), CCAAT/enhancer binding protein alpha (CEBPA), additional sex combs-like 1 (ASXL1), etc.
Patients with MS were followed up through inpatient records, outpatient visits, and telephone calls, with a deadline of April 1, 2022. Overall survival (OS) was calculated as the time from diagnosis of MS until death or last follow-up. Event-free survival (EFS) was defined as the duration between diagnosis to the occurrence of events such as relapse, progression, death for any reason, or last follow-up.
Descriptive statistics were performed for clinicopathological, molecular characteristics, and radiologic attributes. The quantitative data were represented by median (numerical range), while the qualitative data were represented by a number of cases (%). OS and EFS were calculated with the Kaplan–Meier method.
The major clinical features of all cases were summarized in Table 1. Eight males and three females with a median age were 7 years (range 1–13 years). All patients had painless masses or occupying lesions located in the skin, orbital region, lymph nodes central nervous system, testis, or mediastinum. Orbital masses showed symptoms of proptosis or vision loss due to compression. Mediastinal masses caused coughs and polypnea. Only two patients (18%) had their first visit to a hematology department while nine (82%) were non-hematology departments.
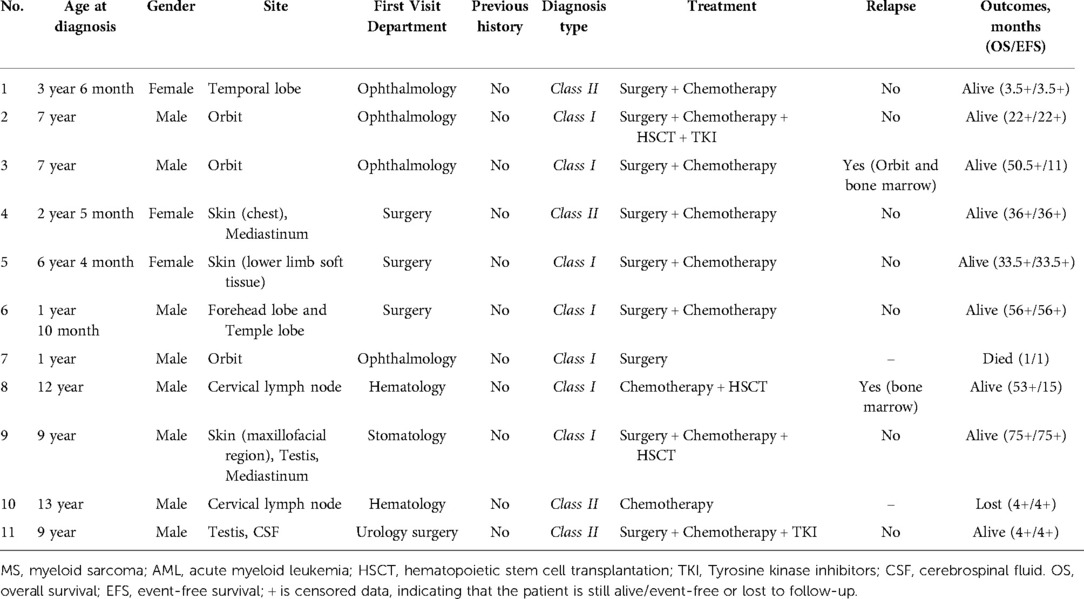
Table 1. Clinical characteristics and outcomes of the patients with MS.
All patients were evaluated by Computed Tomography (CT) or Magnetic Resonance Imaging (MRI) to identify the lesion site. Multi-site involvement in four patients (36%) and single-site in seven (64%). CT showed soft tissue density foci with uneven signal and irregular boundary in most patients. Bone necrosis was seen in some patients. MRI had no specific manifestations. T1-weighted images were mostly showed an equal or low signal, while T2-weighted mainly low signal, enhanced after contrast injection. Radiological data on part patients is shown in Figure 1.
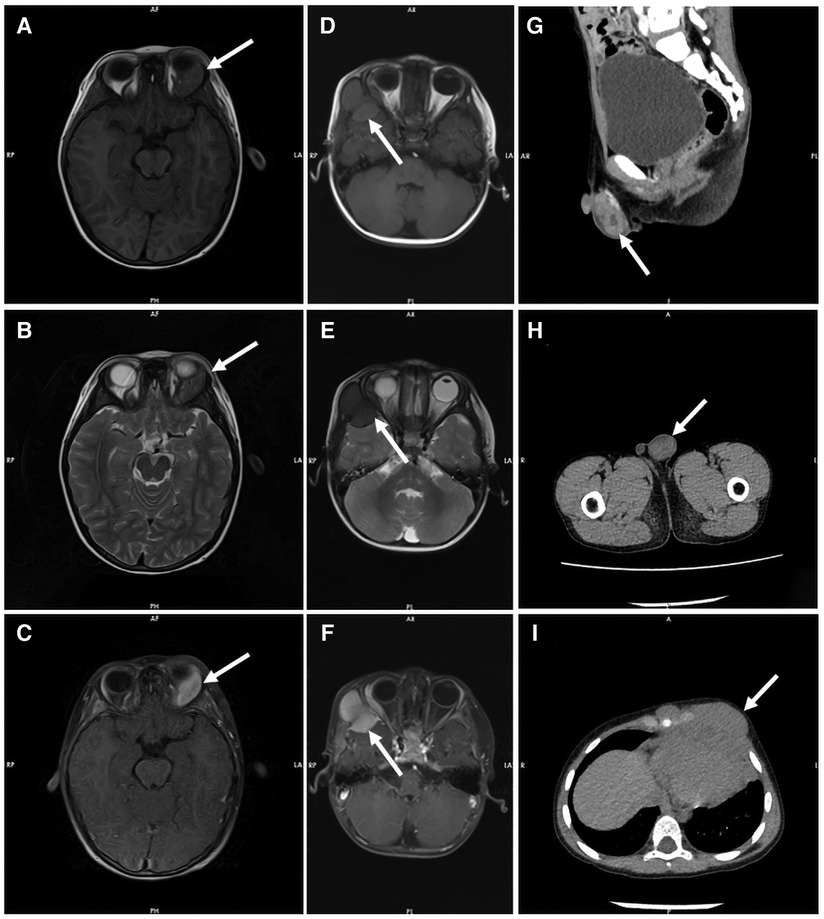
Figure 1. Radiological data on part patients. Both T1-weighted MR images (A) and T2-weighted MR images (B) showed equal signal intensity in the left orbital region of patient 2, which was significantly enhanced by contrast agent injection (C). T1 weighted MR images showed equal signal intensity (D), and T2 weighted MR images showed low signal intensity (E) in the frontal and temporal lobes of patient 6, with uniform enhancement and adjacent brain parenchymal compression (F). CT showed mass soft tissue density foci from the testis of patient 11 (G and H) and the mediastinum of patient 9 (I).
Laboratory data is found in Table 2. Three patients (27%) had anemia or thrombocytopenia at MS diagnosis. Four patients (36%) had leukocytosis and positive blasts in peripheral blood. One patient with positive CSF blasts (Figure 2). Seven patients (64%) were Class I-MS because of notable evidence of AML, with four AML-M2 (36%) and three AML-M5 (27%). Four patients (36%) presented as Class II-MS due to normal bone marrow, without a prior history of the hematopoietic disorder. No patients with Class III and IV-MS in our cohort. Patients diagnosed with AML underwent bone marrow flow cytometry. Six (55%) were positive for classic myeloid makers on flow cytometry, including MPO, CD33, CD34, and CD117. CSF flow cytometry in a patient with isolated MS showed positive myeloid markers for CD33, CD117, CD38, and CD64.
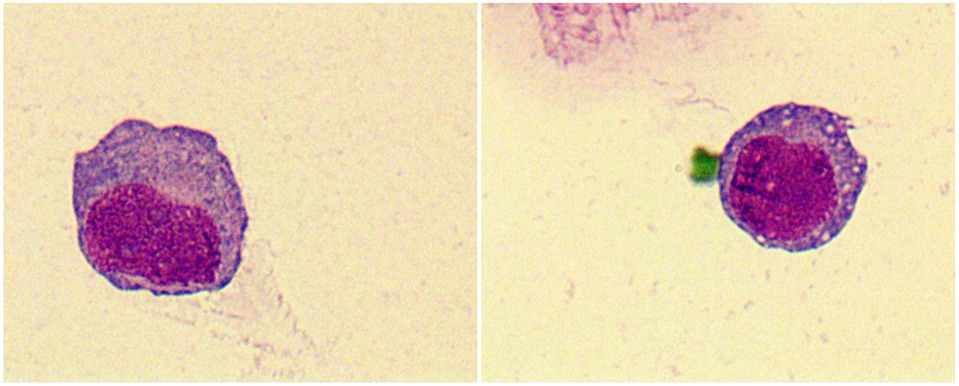
Figure 2. Tumor cells were found in the cerebrospinal fluid (CSF) of patient 11, which have large cytosol and a high amount of envelope. Vacuoles, fine granules and pseudopods were observed in partial cells. The nucleus was large and visibly folded and distorted. Original magnification: 1000×.
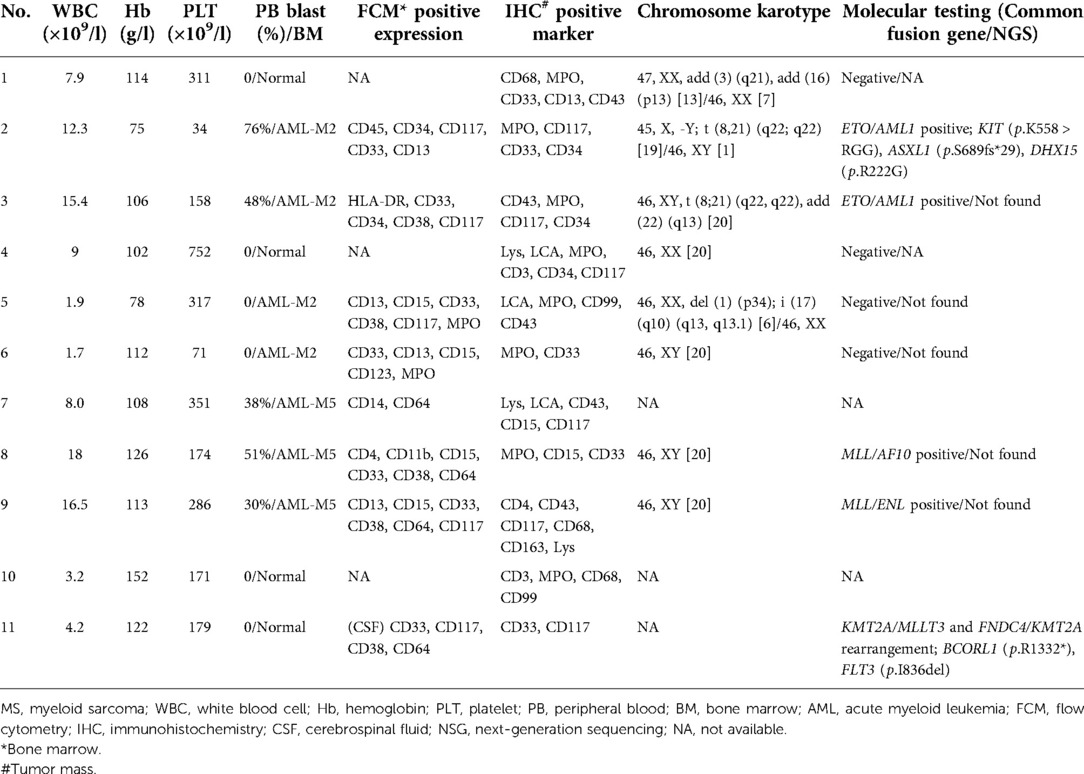
Table 2. Laboratorial, pathological and genetic characteristics of the patients with MS.
Pathologic characteristics were presented in Table 2. On hematoxylin and eosin (H & E) staining, MS was characterized by diffuse infiltration of tissues with myeloid cells, a large nucleus, and irregular nuclear contours. Immunohistochemically, MPO (73%) and CD117 (55%) was the most commonly expressed marker, followed by CD33 (45%), CD43 (45%), CD34 (27%), CD68 (27%), and lysozyme (27%). Patient 11 was notable and fell into a diagnostic dilemma because of limited antibody panels and MPO negative. He was not diagnosed with MS until the genetic results of KMT2A (MLL) gene rearrangement, a common genetic abnormality of AML, were reported, and an expanded antibody panel retested IHC showed positive for CD33 and CD117 (Figure 3).
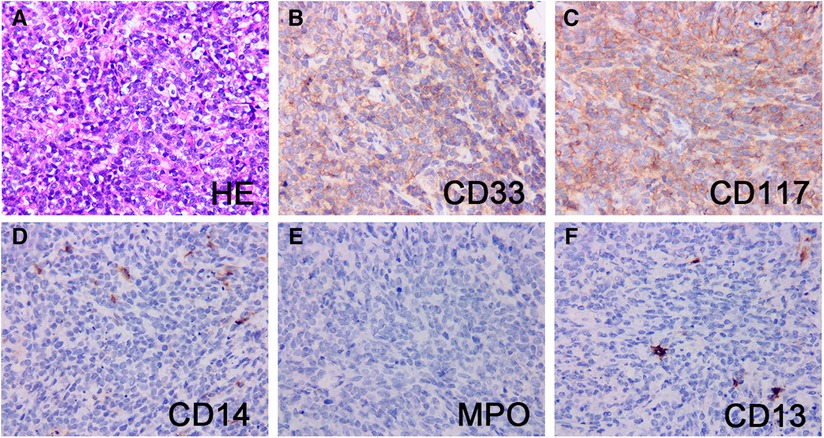
Figure 3. Pathological and immunohistochemical findings of patient 11. H&E stain of a testis mass (A) showed mainly small round cells with abundant cytoplasm, large, irregular nuclei, and easily visible nuclear schizophrenic phase. The neoplastic cells strongly expressed CD33 (B) and CD117 (C), and the blasts were largely negative for CD14 (D), MPO (E) and CD13 (F). Original magnification: 400×.
Karyotyping results were available in eight patients because isolated MS was not routinely examined and one patient with AML refused. Cytogenetic abnormalities were found in four patients (36%) including +3, + 16, + 22, t (8;21), i (17) and del (1). Two patients with t (8; 21) (q22; q22) formed an AML1/ETO fusion gene. Despite normal karyotype, two patients were found positive for MLL/AF10 fusion gene and MLL/ENL, respectively. NGS detected mutations in one MS tissue sample, which showed positive for the KMT2A/MLLT3 and FNDC4/KMT2A gene rearrangement, as well as mutations of BCORL1 (p.R1332*), FLT3 (p.I836del). The bone marrow myeloid mutation panel was identified positively in one patient with KIT (p.K558 > RGG), ASXL1 (p.S689fs*29), and DHX15 (p.R222G).
Nine patients received surgical treatment. All patients were treated with chemotherapy based on AML regimens except one who eventually died of disease progression. AML regimens consisting of induction therapy with daunorubicin and cytarabine and consolidation therapy with high-dose cytarabine. CNS-MS patients were managed with central nervous system leukemia (CNSL) regimen. Eight patients (73%) achieved complete remission (CR) after the first or second induction chemotherapy. Of the eight patients, one patient discontinued chemotherapy and relapsed after 4 months w hile one lost to follow-up. Three patients who achieved CR to induction completed hematopoietic stem cell transplantation (HSCT). Of the three patients, two patients achieved sustained remission, and one relapsed 1 year after. One patient was treated with the addition of dasatinib due to the mutation of the KIT gene, and another patient with sorafenib for the mutation of the FLT3 gene. The median follow-up time was 33.5 months in eleven patients. two patients relapsed, one died, and one lost to follow-up. The 2-year OS rate estimated by Kaplan-Meier curves was 90.9% ± 8.7%, and the EFS rate was 64.9% ± 16.7%.
In this study, we investigated a series of pediatric patients with MS at a chinese single institution. We highlights a precise pathological diagnosis based on immunohistochemical staining and the importance of genetic examination as well as early and regular systemic chemotherapy.
According to the single-center data of 11 children diagnosed with MS, the most common subtype was MS accompanied by AML, which supported the previous literature (9–11). Approximately 6.7%–12% of childhood AML patients had MS at diagnosis (12–15). In fact, the incidence is often underestimated because screening for MS in AML patients is not routinely performed in clinical workups. In our cohort, the incidence of MS with AML may be higher because we only included biopsy-proven MS. We also observed 4 patients with isolated MS, a subtype considered to be less frequent in children. MS can mimic many other systemic diseases due to the variety of symptoms caused by the different sites. In this study, the most common sites were the skin, orbital region, and lymph nodes. Patients with orbital MS mostly presented with AML M2 and M5 subtypes, and AML M5 subtype have a higher initial white blood cell count. These are consistent with previous reports (3, 5, 9, 11).
Radiological test plays an important role in the early recognition of MS. CT and MRI imaging can assess the size and location of the tumor and help distinguish it from other lesions such as hematomas or abscesses (16). Consistent with the literature, T1-weighted images were mainly low signal and equal signal, and the lesions were enhanced with different degrees (17). The difference is that the T2-weighted images of patients in our cohort mostly present equal or low signals. Assessment of the efficacy of extramedullary lesions by imaging is also important. However, there is no standard for the time node and frequency of radiological evaluation. Due to the multi-site involvement of MS, some scholars suggested that positron emission tomography/computed tomography (PET/CT) should be the first choice for the imaging evaluation (18–20).
MS, especially isolated MS, is often a challenging diagnosis in pediatric patients (21). Studies have reported misdiagnosis rates as high as 25% to 47% for isolated MS, and inadequate immunohistochemistry is the most common cause (22). In some cases, the diagnosis of MS is not corrected until a bone marrow biopsy confirms myeloid neoplasms. The panel of antibodies for the diagnosis of MS includes MPO, CD14, CD68, lysozyme, CD117, CD11c, CD13, CD33 (23). MPO, CD117, CD13, CD33 reflect myeloid differentiation, while CD68, CD163, CD14, CD11c are expressed in monocytes (4, 24). The addition of CD20, CD79a, CD3 and CD45RO to the antibody panel excludes B-cell and T-cell lymphomas (3, 25). All cases in this study were positive for at least one antigen reflecting myeloid differentiation or monoclear differentiation, with the highest rates of MPO and CD117. MPO is the most commonly used antibody for the diagnosis of MS, with high sensitivity and specificity. However, it is not expressed in MS tumor cells with low differentiation (26). One patient in our cohort was initially diagnosed with another cancer, which also supported the need for a broad antibody group in immunohistochemistry.
Cytogenetic and molecular biological tests play an important role in the ancillary diagnosis, risk stratification and prognostic assessment of MS. Chromosomal abnormalities in 32%–84% of MS subjects (10, 21). The common genetic abnormalities including t (8;21), inv (16)/t (16;16) or t (15;17), which usually suggested a good prognosis (9, 10, 27). KMT2A (MLL) gene rearrangements or fusions, chromosome 5 or 7 monosomy, 5q-, and complex karyotypes of chromosomes (three or more chromosome aberrations, except good karyotypes) suggested a poor prognosis and high risk (28–30). Patients with positive AML1-ETO fusion gene often presented with orbital masses, while patients with abnormal KMT2A (MLL) gene were characterized by leukocytosis, CNS disease, and testicular involvement, which is consistent with the literature (10, 28). NGS of tumor mass may help identify relevant mutated genes in patients with isolated MS to aid in diagnosis, guide prognosis and treatment, like patient 11.
The prognostic data on the involved sites are different in the literature (29). Johnston et al. showed that pediatric patients with orbital MS and CNS MS presented a lower relapse rate and better outcome than those without extramedullary manifestations (15). Studies suggested that non-skin extramedullary involvement is a favorable prognostic factor for pediatric AML patients (12, 31). In our cohort, one patient with orbital MS had recurrent orbital MS and bone marrow involvement, three patients with skin MS had a good prognosis. Similarly, long-term survival also observed in 2 of 3 skin MS patients in a cohort in Japan (32). Hence, may be more powerful factors, such as molecular characteristics of cancer cells and regular treatments, that determine the prognosis.
Based on previous reports, the treatment of MS is generally systemic chemotherapy, surgery, radiotherapy, HSCT, or a combination of these approaches. Surgery and radiotherapy cannot improve the overall survival of patients but are required when tumors cause obstruction or compression, or leukemia-related skin pain (2, 3, 33). Consistent with the literature (33), patients who received early chemotherapy had significantly longer survival times compared to those who did not receive chemotherapy. Most studies suggested that HSCT after complete remission with chemotherapy improves survival, especially for those who with unfavorable prognostic factors (site, genetic abnormalities, etc.) (2, 3, 9). Two patients in our cohort underwent HSCT after chemotherapy and survived disease free for 22 and 75 months, which also illustrates the importance of HSCT treatment. Targeted agents to improve MS prognosis were also frequently reported such as tyrosine kinase inhibitors (TKI), DNA methyltransferase inhibitors, and CD33 monoclonal antibodies (34, 35). Enhancing the molecular genetic testing of MS patients and implementing individualized treatment may be the future trend.
In summary, MS show a variety of clinical manifestations that can be easily misdiagnosed. Adequate tumor biopsy and immunohistochemistry are necessary for the correct diagnosis of MS. Early and regular systemic chemotherapy promises long-term survival. Due to the rarity of MS, we reviewed too few patients to meaningfully study the impact of potential prognostic factors. It is hoped that future multicenter or even global collaborative large-sample prospective clinical trials will investigate pediatric MS in greater depth.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
FY: data collection, analysis, and interpretation, original draft preparation. HZ: data collection, analysis and interpretation and resources. WZ, JD and WD: investigation. LY: analysis and interpretation, manuscript revision and final approval. All authors have read and agreed to the published version of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (No. 82000137, No. 81770178), and the Natural Science Foundation of Hunan Province of China (No. 2020JJ4918).
We thank the patients and their parents for their participation in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the world health organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
2. Bakst RL, Tallman MS, Douer D, Yahalom J. How I treat extramedullary acute myeloid leukemia. Blood. (2011) 118:3785–93. doi: 10.1182/blood-2011-04-347229
3. Avni B, Koren-Michowitz M. Myeloid sarcoma: current approach and therapeutic options. Ther Adv Hematol. (2011) 2:309–16. doi: 10.1177/2040620711410774
4. Klco JM, Welch JS, Nguyen TT, Hurley MY, Kreisel FH, Hassan A, et al. State of the art in myeloid sarcoma. Int J Lab Hematol. (2011) 33:555–65. doi: 10.1111/j.1751-553X.2011.01361.x
5. Ohanian M, Faderl S, Ravandi F, Pemmaraju N, Garcia-Manero G, Cortes J, et al. Is acute myeloid leukemia a liquid tumor? Int J Cancer. (2013) 133:534–43. doi: 10.1002/ijc.28012
6. Wilson CS, Medeiros LJ. Extramedullary manifestations of myeloid neoplasms. AM J Clin Pathol. (2015) 144:219–39. doi: 10.1309/AJCPO58YWIBUBESX
7. Almond LM, Charalampakis M, Ford SJ, Gourevitch D, Desai A. Myeloid sarcoma: presentation, diagnosis, and treatment. Clin Lymphoma Myeloma Leuk. (2017) 17:263–7. doi: 10.1016/j.clml.2017.02.027
8. Polyatskin IL, Artemyeva AS, Krivolapov YA. [Revised WHO classification of tumors of hematopoietic and lymphoid tissues, 2017 (4th edition):lymphoid tumors]. Arkh Patol. (2019) 81:59–65. doi: 10.17116/patol20198103159
9. Zhou T, Bloomquist MS, Ferguson LS, Reuther J, Marcogliese AN, Elghetany MT, et al. Pediatric myeloid sarcoma: a single institution clinicopathologic and molecular analysis. Pediatr Hematol Oncol. (2020) 37:76–89. doi: 10.1080/08880018.2019.1683107
10. Hu G, Lu A, Wu J, Jia Y, Zuo Y, Ding M, et al. Characteristics and prognosis of pediatric myeloid sarcoma in the cytogenetic context of t(8;21). Pediatr Hematol Oncol. (2021) 38:14–24. doi: 10.1080/08880018.2020.1803462
11. Samborska M, Baranska M, Wachowiak J, Skalska-Sadowska J, Thambyrajah S, Czogala M, et al. Clinical characteristics and treatment outcomes of myeloid sarcoma in children: the experience of the Polish pediatric leukemia and lymphoma study group. Front Oncol. (2022) 12:935373. doi: 10.3389/fonc.2022.935373
12. Dusenbery KE, Howells WB, Arthur DC, Alonzo T, Lee JW, Kobrinsky N, et al. Extramedullary leukemia in children with newly diagnosed acute myeloid leukemia: a report from the Children's Cancer group. J Pediatr Hematol Oncol. (2003) 25:760–8. doi: 10.1097/00043426-200310000-00004
13. Kobayashi R, Tawa A, Hanada R, Horibe K, Tsuchida M, Tsukimoto I. Extramedullary infiltration at diagnosis and prognosis in children with acute myelogenous leukemia. Pediatr Blood Cancer. (2007) 48:393–8. doi: 10.1002/pbc.20824
14. Stove HK, Sandahl JD, Abrahamsson J, Asdahl PH, Forestier E, Ha SY, et al. Extramedullary leukemia in children with acute myeloid leukemia: a population-based cohort study from the nordic society of pediatric hematology and oncology (NOPHO). Pediatr Blood Cancer. (2017) 64(12):e26520. doi: 10.1002/pbc.26520
15. Johnston DL, Alonzo TA, Gerbing RB, Lange BJ, Woods WG. Superior outcome of pediatric acute myeloid leukemia patients with orbital and CNS myeloid sarcoma: a report from the Children's Oncology group. Pediatr Blood Cancer. (2012) 58:519–24. doi: 10.1002/pbc.23201
16. Yilmaz AF, Saydam G, Sahin F, Baran Y. Granulocytic sarcoma: a systematic review. Am J Blood Res. (2013) 3(4):265–70. PMID: 24396704 PMCID: PMC387527524396704
17. Meyer HJ, Ponisch W, Schmidt SA, Wienbeck S, Braulke F, Schramm D, et al. Clinical and imaging features of myeloid sarcoma: a German multicenter study. BMC Cancer. (2019) 19:1150. doi: 10.1186/s12885-019-6357-y
18. He HS, Su GP, Yao JP, Liu SH, Xu YH, Yang YQ, et al. [Clinical characteristics of patients with myeloid sarcoma]. Zhongguo Shi Yan Xue Ye Xue Za Zhi. (2020) 28:1491–5. doi: 10.19746/j.cnki.issn.1009-2137.2020.05.011
19. Bakst R, Powers A, Yahalom J. Diagnostic and therapeutic considerations for extramedullary leukemia. Curr Oncol Rep. (2020) 22:75. doi: 10.1007/s11912-020-00919-6
20. Stolzel F, Luer T, Lock S, Parmentier S, Kuithan F, Kramer M, et al. The prevalence of extramedullary acute myeloid leukemia detected by (18)FDG-PET/CT: final results from the prospective PETAML trial. Haematologica. (2020) 105:1552–8. doi: 10.3324/haematol.2019.223032
21. Audouin J, Comperat E, Le Tourneau A, Camilleri-Broet S, Adida C, Molina T, et al. Myeloid sarcoma: clinical and morphologic criteria useful for diagnosis. Int J Surg Pathol. (2003) 11:271–82. doi: 10.1177/106689690301100404
22. Seifert RP, Bulkeley WR, Zhang L, Menes M, Bui MM. A practical approach to diagnose soft tissue myeloid sarcoma preceding or coinciding with acute myeloid leukemia. Ann Diagn Pathol. (2014) 18:253–60. doi: 10.1016/j.anndiagpath.2014.06.001
23. Pileri SA, Ascani S, Cox MC, Campidelli C, Bacci F, Piccioli M, et al. Myeloid sarcoma: clinico-pathologic, phenotypic and cytogenetic analysis of 92 adult patients. Leukemia. (2007) 21:340–50. doi: 10.1038/sj.leu.2404491
24. Alexiev BA, Wang W, Ning Y, Chumsri S, Gojo I, Rodgers WH, et al. Myeloid sarcomas: a histologic, immunohistochemical, and cytogenetic study. Diagn Pathol. (2007) 2:42. doi: 10.1186/1746-1596-2-42
25. Campidelli C, Agostinelli C, Stitson R, Pileri SA. Myeloid sarcoma: extramedullary manifestation of myeloid disorders. Am J Clin Pathol. (2009) 132:426–37. doi: 10.1309/AJCP1ZA7HYZKAZHS
26. Suh YK, Shin HJ. Fine-needle aspiration biopsy of granulocytic sarcoma: a clinicopathologic study of 27 cases. Cancer-Am Cancer Soc. (2000) 90(6):364–72.
27. Zhang XH, Zhang R, Li Y. Granulocytic sarcoma of abdomen in acute myeloid leukemia patient with inv(16) and t(6;17) abnormal chromosome: case report and review of literature. Leuk Res. (2010) 34:958–61. doi: 10.1016/j.leukres.2010.01.009
28. El JS, Salama A, Marcellino BK, Abulsayen HA, Zhou X, Hassan M, et al. Myeloid sarcoma of the testis in children: clinicopathologic and immunohistochemical characteristics with KMT2A (MLL) gene rearrangement correlation. Appl Immunohistochem Mol Morphol. (2020) 28:501–7. doi: 10.1097/PAI.0000000000000783
29. Xu LH, Wang Y, Chen ZY, Fang JP. Myeloid sarcoma is associated with poor clinical outcome in pediatric patients with acute myeloid leukemia. J Cancer Res Clin Oncol. (2020) 146:1011–20. doi: 10.1007/s00432-020-03128-7
30. Park KU, Lee DS, Lee HS, Kim CJ, Cho HI. Granulocytic sarcoma in MLL-positive infant acute myelogenous leukemia: fluorescence in situ hybridization study of childhood acute myelogenous leukemia for detecting MLL rearrangement. Am J Pathol. (2001) 159:2011–6. doi: 10.1016/S0002-9440(10)63052-0
31. Cho-Vega JH, Medeiros LJ, Prieto VG, Vega F. Leukemia cutis. Am J Clin Pathol. (2008) 129:130–42. doi: 10.1309/WYACYWF6NGM3WBRT
32. Taga T, Imamura T, Nakashima K, Maeda N, Watanabe A, Miyajima Y, et al. Clinical characteristics of pediatric patients with myeloid sarcoma without bone marrow involvement in Japan. Int J Hematol. (2018) 108:438–42. doi: 10.1007/s12185-018-2492-5
33. Lan TY, Lin DT, Tien HF, Yang RS, Chen CY, Wu K. Prognostic factors of treatment outcomes in patients with granulocytic sarcoma. Acta Haematol. (2009) 122:238–46. doi: 10.1159/000253592
34. Ando T, Mitani N, Matsunaga K, Nakazora T, Gondo T, Yujiri T, et al. Gemtuzumab ozogamicin therapy for isolated extramedullary AML relapse after allogeneic hematopoietic stem-cell transplantation. Tohoku J Exp Med. (2010) 220:121–6. doi: 10.1620/tjem.220.121
Keywords: pediatric, myeloid sarcoma, clinical characteristics, pathology, acute myeloid leukemia
Citation: Ye F, Zhang H, Zhang W, Dong J, Deng W and Yang L (2022) Clinical characteristics, pathology features and outcomes of pediatric myeloid sarcoma: A retrospective case series. Front. Pediatr. 10:927894. doi: 10.3389/fped.2022.927894
Received: 25 April 2022; Accepted: 16 November 2022;
Published: 5 December 2022.
Edited by:
Maurizio Aricò, University of Chieti-Pescara, ItalyReviewed by:
Anna Maria Testi, Sapienza University of Rome, Italy© 2022 Ye, Zhang, Zhang, Dong, Deng and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liangchun Yang eWFuZ2xpYW5nY2h1bmdAMTYzLmNvbQ==
Specialty Section: This article was submitted to Pediatric Hematology and Hematological Malignancies, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.