 Zhenyu Liao
Zhenyu Liao Yali Liu2†
Yali Liu2† Qin Lu
Qin Lu Yu Peng
Yu Peng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 13 July 2022
Sec. Pediatric Neurology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.927392
Background: The NALCN encodes a sodium ion leak channel that regulates nerve-resting conductance and excitability. NALCN variants are associated with two neurodevelopmental disorders, one is CLIFAHDD (autosomal dominant congenital contractures of the limbs and face, hypotonia, and developmental delay, OMIM #616266) and another is IHPRF (infantile hypotonia with psychomotor retardation, and characteristic facies 1, OMIM #615419).
Case Presentation: In the current study, a Chinese infant that manifested abnormal facial features, adducted thumbs, and neurodevelopmental retardation was diagnosed with CLIFAHDD syndrome. A trio-based whole-exome sequencing revealed that the infant harbored a de novo variant of the NALCN gene (c.4300A>G, p.I1434V).
Conclusions: Our findings further enriched the variant spectrum of the NALCN gene and may expand the clinical range of NALCN-related disorders.
The NALCN encodes a voltage-independent, non-selective cation channel (1, 2) expressed mainly in the central nervous system (CNS) (3–6). NALCN plays a significant role in mediating a background Na+ leak current (IL-NA) that determines the nerve-resting conductance and excitability of different types of cells (including neurons). For instance, NALCN forms a sodium channel complex with UNC80 and UNC79 (7) and is involved in many physiological processes, such as respiration (8), intestinal pacing (9), and circadian rhythm cycle regulation (10).
The NALCN variants were associated with various diseases, such as IHPRF (infantile hypotonia with psychomotor retardation and characteristic facies, OMIM #615419) (11), and CLIFAHDD (congenital contractures of the limbs and face, hypotonia, and developmental delay, OMIM #616266) (12, 13). CLIFAHDD syndrome was first reported by 10(12). It is characterized by congenital contractures of the limbs and face, hypotonia, and developmental delay inherited in an autosomal dominant manner (12, 14). To date, all the published NALCN variants of patients with CLIFAHDD have occurred de novo to our knowledge (12, 13). IHPRF is characterized by severe hypotonia, psychomotor retardation, and characteristic facies at birth, or early infancy inherited in an autosomal recessive manner (15). Both CLIFAHDD and IHPRF were neurodevelopmental diseases, although they shared some characteristics (e.g., developmental delay and hypotonia), they are otherwise distinct from one another. Their major differences are whether they have distal joint contracture and different inheritance patterns (12, 16, 17).
This study identified a novel NALCN variant (NM_052867.3, c.4300A>G) in a Chinese patient with CLIFAHDD. Our findings further enriched the variant spectrum of the NALCN gene and may expand the clinical range of NALCN-related disorders.
The DNA was isolated from peripheral blood. The Novaseq 6000 platform (Illumina, San Diego, USA) with 150 bp pair-end reads was applied for sequencing. The general procedure for whole-exome sequencing (WES) was as follows: genomic DNA was sheared to a fragment size of around 150 bp and with blunt-end, followed by the addition of deoxyadenosine at the 3'ends of the fragments. The genomic DNA library was created by ligating adaptors to the ends of double DNA strands that enable sequencing. The library was amplified by PCR and then, hybridized into a pool of biotinylated oligo probes specific for exons. Streptavidin magnetic beads were used to capture DNA-probe hybrids. Raw image files were processed through CASAVA v1.82 for base calling and generating raw data with adequate consensus coding sequences (CCDS) coverages (93.93%-94.96%, for depth ≥ 20).
Burrows-Wheeler Aligner tool was used to map the sequencing reads to the human reference genome (hg19/GRCh37), and PCR duplicates were removed by Picard v1.57 (http://picard.sourceforge.net/). Variation annotation was conducted according to the American College of Medical Genetics and Genomics (ACMG) guidelines (18) and the Enliven® Variants Annotation Interpretation System authorized by Berry Genomics. The parent-child relationship was identified by King software (v2.2.5) (19) to confirm the de novo to confirm the de novo origin of the variant.
Sanger sequencing was used to validate the variant identified by WES. Primers were designed by the Primer3 program (https://primer3.ut.ee/).
A 3-day-old boy was admitted to the hospital for 24 h due to jaundice. At 38 weeks of natural pregnancy, he was born to a 30-year-old pregnant woman, G1P1. There was no abnormality except for the cord around the neck during the entire term. The baby weighed 3,100 g, the height was 47 cm and the occipito-frontal circumference was 34.5 cm at birth, Apgar scored ten at 1 min, and five at 10 min. His parents are non-consanguineous without a family history of genetic disease.
Physical examination showed that the head, face, and trunk skin had moderate jaundice. Facial features included down slanting palpebral fissures, broad nasal bridge, large nares, long philtrum, micrognathia, pursed lips, H-shaped chin dimpling, deep nasolabial folds, and full cheeks were observed (Figure 1A). He also had an unusually short neck, adducted thumbs, clinodactyly of fingers, transverse palmar areas, elbow contracture, and toe deformity (Figures 1B,C). Anterior fontanelle (1.5*1.5 cm), pupillary morphology, and light sensitivity were normal. There was no apparent abnormality in the oral cavity, pharynx, lungs, chest, abdomen, anus, external genitalia, muscle tension of limbs, etc.
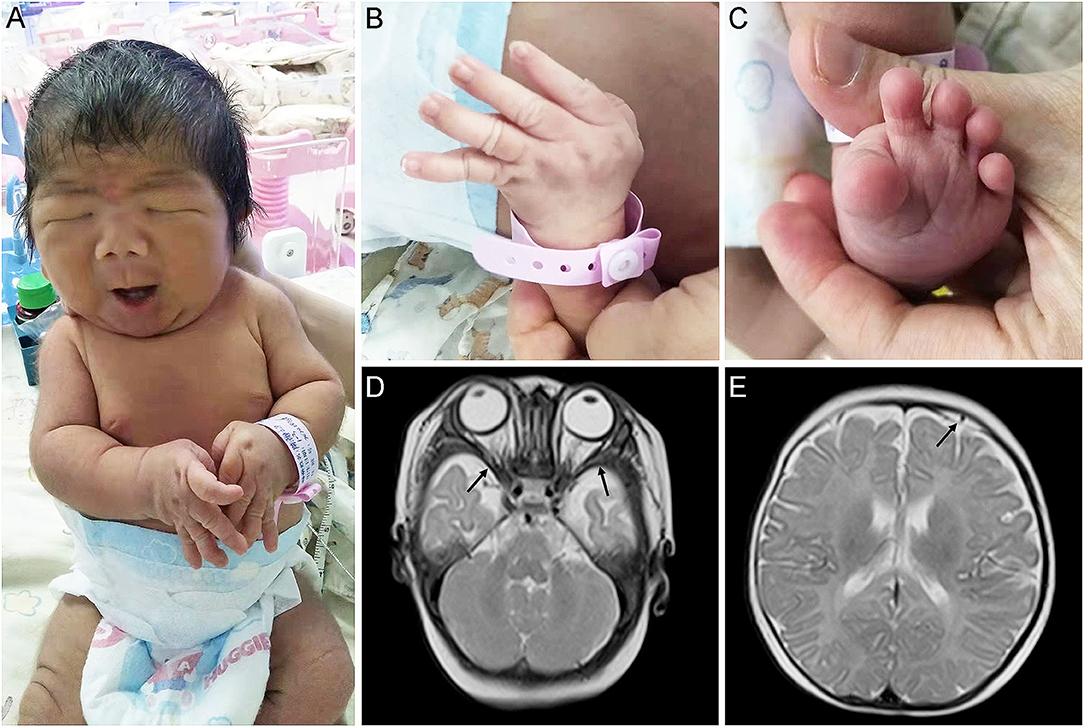
Figure 1. Appearance and MRI features of the proband. (A,B) Congenital contractures of the face (A) and limbs (B). (C) Deformity of the foot. (D) abnormally broadening of bilateral temporal extracranial space. (E) Subdural effusion in the left frontal area.
The MRI revealed that he has abnormally broadening bilateral temporal extracranial space (Figure 1D), left frontal subdural effusion (Figure 1E), and cyst in the right choroid plexus (3*2 mm). In addition, a color doppler ultrasound found that there was a patent foramen ovale in his heart (2.8 mm). His blood biochemical profile showed that he suffered from indirect hyperbilirubinemia, but the liver and kidney function was normal. Also, he suffered from anemia. After receiving phototherapy for jaundice, the infant's condition improved. Then, his parents decided to discharge him from the hospital.
Three months later, a physical examination showed that the infant manifested neurodevelopmental delay, unable to finish the Bailey Infant Development Scale. Moreover, his neonatal behavioral neurological assessment (NBNA) was only 31 points. The performance was observed as follows: no response of his head and eyes to the gurgling sound, the red ball, and the speaking face; incomplete support response at the upright position, grasp weakness, poor stretch reflection with only part of the body lifted, challenging to trigger the stepping reflex, and incomplete Moro reflex. At present, the proband is two years and six months old, and he is unable to speak his first words and he has no response to the orders with a score of 25 accessed by Gesell Developmental Schedules. However, he can still walk without fine motor control. His parents refused reexamination of the head by MRI and only took free rehabilitation training.
A de novo missense variant was identified in the NALCN gene (NM_052867.3: c.4300A>G, p.I1434V) by WES. The relationship between the three samples was confirmed (Supplementary Table S2). Sanger sequencing confirmed that the variant was heterozygous in the affected child but was absent from his parents (Figure 2). The variant was not found in gnomAD exomes or gnomAD genomes (http://gnomad.broadinstitute.org). In addition, the gnomAD missense Z score of the NALCN gene was 4.96 (> 3.09), and the assessment using multiple computational in silico tools indicated that the identified substitution was pathogenic or damaging (8 pathogenic vs. 3 benign predictions). Overall, the ACMG classification of the variant was likely pathogenic (PS2, PM2, PP2, and PP3).
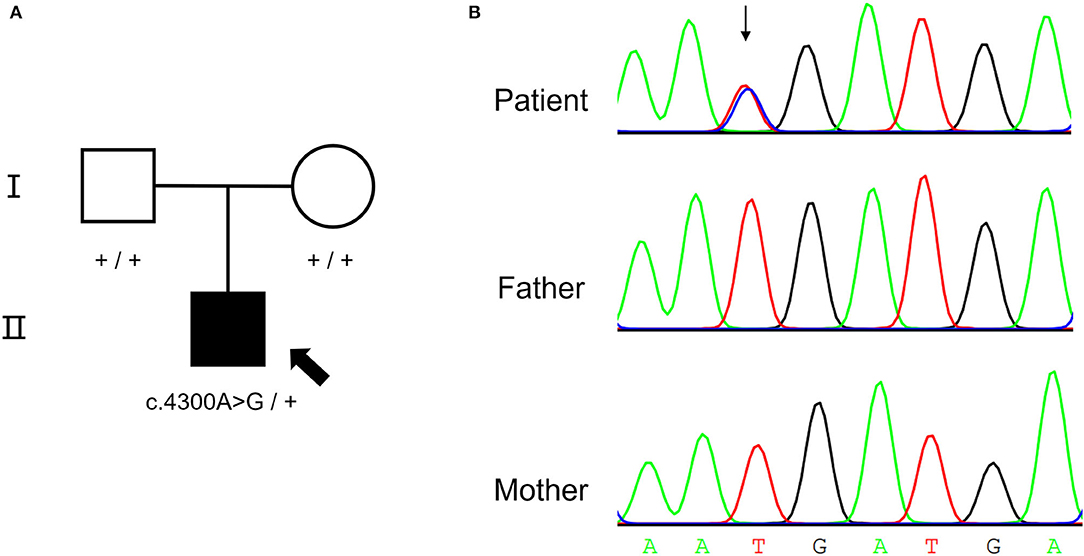
Figure 2. De novo NALCN variant of the proband. (A) The pedigree of the CLIFAHDD family. (B) Sanger sequencing result shows c.4300A>G in NALCN gene.
The CLIFAHDD was first reported by Chong et al. (12). In this study, we reported a CLIFAHDD infant with a NALCN gene variant (c.4300A>G, p.I1434V). Besides, we found a cyst in the right choroid plexus (3*2 mm) in our patient, which was also found in the patient with the pathogenic variant p.Y578C (13). Cyst in the choroid plexus could be detected in 8.8% of neonates, and most of the cyst could be resolved spontaneously, the association between the cyst and developmental delay had not been built (20). At present, the status of the cyst in our case was not available, as the parents refused further examination.
The NALCN gene contains 1,738 amino acids and 44 exons, located on chromosome 13q33.1 (1). NALCN is a voltage-independent, non-selective cationic channel, which has four homologous repeats (I-IV), each composed of six transmembrane segments (S1–S6) (21, 22). The sodium leakage currents are transferred through the pore of the channel, four pore-forming loops (P-loops) spanning from S5 to S6 making up the ion selectivity filter, which is essential for controlling the resting and conducting membrane potential through persistent sodium leakage currents, contributing to nervous tension excitatory (21, 23). There were 23 de novo NALCN variants, which were reported to be associated with CLIFAHDD or distal arthrogryposis (6, 12–14, 16, 24–26), all were located in or near the S5 and S6 segments, and the clinical spectrum was reviewed (Supplementary Table S1). Previous reports indicated that facial contractures, arthrogryposis, joint contractures, hypotonia, and development delay (including cognitive delay, motor delay, and speech delay) were among the most prevalent manifestations; cerebellar atrophy was the most common abnormality found by MRI; although other clinical features varied. There were two previously reported variants (p.I1445L and p.I1446M) located in the S6 of the fourth domain, the patient with the p.I1445L variant presented no arthrogryposis and joint contractures (26), while the patient with p.I1446M variant presented no cerebellar atrophy (12). In our case, the proband displayed no hypotonia but suffered from facial contractures, arthrogryposis, and development delay. Our findings supported the high variability of the clinical phenotype caused by de novo NALCN missense variants. To date, there were no efficient treatments for CLIFAHDD, except for symptomatic treatments, such as clonazepam for seizures (24), acetazolamide for episodic ataxia (16), and surgical procedures for arthrogryposis (27). A 2-aminoethoxydiphenyl borate and a flunarizine could rescue the hyperactive phenotype of C. elegans caused by NALCN gain-of-function variants or it might serve as a direct blocker of the channel (28). However, further validation was needed for clinical implementation.
Contrarily, biallelic loss-of-function variants of NALCN often led to the IHPRF syndrome (15, 17, 29). In addition, the parents of patients with IHPRF were carriers of NALCN variants but reportedly did not show any symptoms of CLIFAHDD syndrome (12). The phenomena suggested that CLIFAHDD and IHPRF are caused by different mechanisms. Research has shown that some of these variants resulted in a dominant gain-of-function defect. For instance, four human CLIFAHDD variants (p.T513N, p.F512V, p.L509S, and p.L590F) in the orthologous position of the C. elegans result in gain-of-function phenotypes, which were previously proven (30), like hypertonia, smaller body size, and curly posture (24). Another research revealed that NALCN channels contribute to an inward Na+ background current in differentiated NG108-15 cells, and the current amplitude was a time-dependent decay; expression of the two CLIFAHDD variants, p.L509S, and p.Y578S, showed Na+ background current of significantly higher current density and a significant slowing of the current inactivation. On the contrary, expression of the IHPRF p.W1287L variant did not result in any detectable Na+ background current (31), indicating a non-functional NALCN channel. These results implied that gain-of-function variants of NALCN might result in CLIFAHDD, while loss-of-function variants might result in IHPRF. Further study was needed to explore the detailed mechanism.
Nevertheless, we also found a de novo BPTF variant (NM_182641.3: c.1117A>G, p.T373A) in this infant. It was classified as a VUS variant according to ACMG criteria (PS2_surpporting+PM2_supporting). A heterozygous variant of BPTF may cause neurodevelopmental disorder with dysmorphic facies and distal limb anomalies (NEDDFL). However, the NEDDFL manifested with abnormalities of the hands and feet, and the facial features are distinct from CLIFAHDD. Thus, we considered that the BPTF variant was not the causal variant.
In conclusion, we first described CLIFAHDD phenotypes associated with a variant of the NALCN gene (c.4300A>G, p.I1434V) in a Chinese infant. Our findings further enriched the variant spectrum of the NALCN gene and may expand the clinical range of NALCN-related disorders.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, 2565796.
The studies involving human participants were reviewed and approved by the Ethics Committee of Hunan Children's Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
ZL, YL, and QLi: supervision, resources acquisition, and original manuscript writing and editing. ZL, QLu, and YW: methodology and genetic experiments. YP: data analysis and manuscript review and editing. All authors read and approved the final manuscript.
This work was supported by a grant from Major Scientific and Technological Projects of Hunan Province for collaborative prevention and control of birth defects: 2019SK1014.
YW was employed by GeneMind Biosciences Company Limited, ShenZhen, China. QLu was employed by GeneTalks Biotech Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors greatly thank all the patients and their family members who participated in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.927392/full#supplementary-material
Supplementary Table S1. The previous reported NALCN variants and phenotypes in CLIFAHDD.
Supplementary Table S2. Relationship confirm by King software.
1. Snutch TP, Monteil A. The sodium “leak” has finally been plugged. Neuron. (2007) 54:505–7. doi: 10.1016/j.neuron.2007.05.005
2. Zhou C, Luo J, He X, Zhou Q, He Y, Wang X, et al. The NALCN channel regulator unc-80 functions in a subset of interneurons to regulate caenorhabditis elegans reversal behavior. G3 (Bethesda). (2020) 10:199–210. doi: 10.1534/g3.119.400692
3. Lee JH, Cribbs LL, Perez-Reyes E. Cloning of a novel four repeat protein related to voltage-gated sodium and calcium channels. FEBS Lett. (1999) 445:231–6. doi: 10.1016/S0014-5793(99)00082-4
4. Kutlu B, Burdick D, Baxter D, Rasschaert J, Flamez D, Eizirik DL, et al. Detailed transcriptome atlas of the pancreatic beta cell. BMC Med Genomics. (2009) 2:3. doi: 10.1186/1755-8794-2-3
5. Swayne LA, Mezghrani A, Varrault A, Chemin J, Bertrand G, Dalle S, et al. The NALCN ion channel is activated by M3 muscarinic receptors in a pancreatic beta-cell line. EMBO Rep. (2009) 10:873–80. doi: 10.1038/embor.2009.125
6. Karakaya M, Heller R, Kunde V, Zimmer KP, Chao CM, Nurnberg P, et al. Novel Mutations in the Nonselective Sodium Leak Channel (NALCN) lead to distal arthrogryposis with increased muscle tone. Neuropediatrics. (2016) 47:273–7. doi: 10.1055/s-0036-1584084
7. Wie J, Bharthur A, Wolfgang M, Narayanan V, Ramsey K, Group CRR, et al. Intellectual disability-associated UNC80 mutations reveal inter-subunit interaction and dendritic function of the NALCN channel complex. Nat Commun. (2020) 11:3351. doi: 10.1038/s41467-020-17105-8
8. Bourque DK, Dyment DA, MacLusky I, Kernohan KD. Care4Rare Canada C, McMillan HJ. Periodic breathing in patients with NALCN mutations. J Hum Genet. (2018) 63:1093–6. doi: 10.1038/s10038-018-0484-1
9. Kim BJ, Chang IY, Choi S, Jun JY, Jeon JH, Xu WX, et al. Involvement of Na(+)-leak channel in substance P-induced depolarization of pacemaking activity in interstitial cells of Cajal. Cell Physiol Biochem. (2012) 29:501–10. doi: 10.1159/000338504
10. Chua HC, Wulf M, Weidling C, Rasmussen LP, Pless SA. The NALCN channel complex is voltage sensitive and directly modulated by extracellular calcium, Sci Adv. (2020) 6: eaaz3154. doi: 10.1126/sciadv.aaz3154
11. Shojaei M, Rashedi S, Chakoli AN, Karimi AH, Geraily G. A homozygous truncating mutation in NALCN Causing IHPRF1: detailed clinical manifestations and a review of literature. Appl Clin Genet. (2020) 13:151–7. doi: 10.2147/TACG.S261781
12. Chong JX, McMillin MJ, Shively KM, Beck AE, Marvin CT, Armenteros JR, et al. De novo mutations in NALCN cause a syndrome characterized by congenital contractures of the limbs and face, hypotonia, and developmental delay. Am J Hum Genet. (2015) 96:462–73. doi: 10.3113/JSOA.2015.0213
13. Vivero M, Cho MT, Begtrup A, Wentzensen IM, Walsh L, Payne K, et al. Additional de novo missense genetic variants in NALCN associated with CLIFAHDD syndrome. Clin Genet. (2017) 91:929–31. doi: 10.1111/cge.12899
14. Fukai R, Saitsu H, Okamoto N, Sakai Y, Fattal-Valevski A, Masaaki S, et al. De novo missense mutations in NALCN cause developmental and intellectual impairment with hypotonia. J Hum Genet. (2016) 61:451–5. doi: 10.1038/jhg.2015.163
15. Al-Sayed MD, Al-Zaidan H, Albakheet A, Hakami H, Kenana R, Al-Yafee Y, et al. Mutations in NALCN cause an autosomal-recessive syndrome with severe hypotonia, speech impairment, and cognitive delay. Am J Hum Genet. (2013) 93:721–26. doi: 10.1016/j.ajhg.2013.08.001
16. Aoyagi K, Rossignol E, Hamdan FF, Mulcahy B, Xie L, Nagamatsu S, et al. A gain-of-function mutation in NALCN in a child with intellectual disability. Ataxia, and Arthrogryposis. Hum Mutat. (2015) 36:753–7. doi: 10.1002/humu.22797
17. Gal M, Magen D, Zahran Y, Ravid S, Eran A, Khayat M, et al. A novel homozygous splice site mutation in NALCN identified in siblings with cachexia, strabismus, severe intellectual disability, epilepsy and abnormal respiratory rhythm. Eur J Med Genet. (2016) 59:204–9. doi: 10.1016/j.ejmg.2016.02.007
18. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
19. Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome-wide association studies. Bioinformatics. (2010) 26:2867–73. doi: 10.1093/bioinformatics/btq559
20. Hung KL, Liao HT. Neonatal choroid plexus cysts and early childhood developmental outcome, J Formos Med Assoc. (2002) 101:43–7.
21. Lu B, Su Y, Das S, Liu J, Xia J, Ren D. The neuronal channel NALCN contributes resting sodium permeability and is required for normal respiratory rhythm. Cell. (2007) 129:371–83. doi: 10.1016/j.cell.2007.02.041
22. Cochet-Bissuel M, Lory P, Monteil A. The sodium leak channel. NALCN, in health and disease. Front Cell Neurosci. (2014) 8:132. doi: 10.3389/fncel.2014.00132
23. Gao S, Xie L, Kawano T, Po MD, Guan S, Zhen M, et al. The NCA sodium leak channel is required for persistent motor circuit activity that sustains locomotion. Nat Commun. (2015) 6:6323. doi: 10.1038/ncomms7323
24. Bend EG, Si Y, Stevenson DA, Bayrak-Toydemir P, Newcomb TM, Jorgensen EM, et al. NALCN channelopathies: distinguishing gain-of-function and loss-of-function mutations. Neurology. (2016) 87:1131–9. doi: 10.1212/WNL.0000000000003095
25. Sivaraman I, Friedman NR, Prayson RA. Muscle biopsy findings in a child with NALCN gene mutation. J Clin Neurosci. (2016) 34 222–23. doi: 10.1016/j.jocn.2016.06.018
26. Bramswig NC, Bertoli-Avella AM, Albrecht B, Al Aqeel AI, Alhashem A, Al-Sannaa N, et al. Genetic variants in components of the NALCN-UNC80-UNC79 ion channel complex cause a broad clinical phenotype (NALCN channelopathies). Hum Genet. (2018) 137:753–68. doi: 10.1007/s00439-018-1929-5
27. Ma L, Yu X. Arthrogryposis multiplex congenita: classification diagnosis, perioperative care, and anesthesia. Front Med. (2017) 11:48–52. doi: 10.1007/s11684-017-0500-4
28. Kasap M, Aamodt EJ, Sagrera CE, Dwyer DS. Novel pharmacological modulation of dystonic phenotypes caused by a gain-of-function mutation in the Na+ leak-current channel, Behav Pharmacol. (2020) 31:465–6. doi: 10.1097/FBP.0000000000000526
29. Koroglu C, Seven M, Tolun A. Recessive truncating NALCN mutation in infantile neuroaxonal dystrophy with facial dysmorphism. J Med Genet. (2013) 50:515–20. doi: 10.1136/jmedgenet-2013-101634
30. Yeh E, Ng S, Zhang M, Bouhours M, Wang Y, Wang M, et al. A putative cation channel, NCA-1, and a novel protein, UNC-80, transmit neuronal activity in C elegans. PLoS Biol. (2008) 6:e55. doi: 10.1371/journal.pbio.0060055
Keywords: NALCN, sodium ion leak channel, CLIFAHDD, neurodevelopmental retardation, de novo mutation
Citation: Liao Z, Liu Y, Wang Y, Lu Q, Peng Y and Liu Q (2022) Case Report: A de novo Variant in NALCN Associated With CLIFAHDD Syndrome in a Chinese Infant. Front. Pediatr. 10:927392. doi: 10.3389/fped.2022.927392
Received: 26 April 2022; Accepted: 13 June 2022;
Published: 13 July 2022.
Edited by:
Velibor Tasic, Saints Cyril and Methodius University of Skopje, North MacedoniaReviewed by:
Adrijan Sarajlija, The Institute for Health Protection of Mother and Child Serbia, SerbiaCopyright © 2022 Liao, Liu, Wang, Lu, Peng and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Peng, ZWluc3RlaW5weUBnbWFpbC5jb20=; Qingsong Liu, MzYxNTM5MDgzQHFxLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.