
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 15 July 2022
Sec. Pediatric Hematology and Hematological Malignancies
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.925599
This article is part of the Research Topic Insights in Thalassemia: From Genomics to Clinical Practice View all 10 articles
 Yetti Hernaningsih1*
Yetti Hernaningsih1* Yuli Syafitri2
Yuli Syafitri2 Yulia Nadar Indrasari1
Yulia Nadar Indrasari1 Prafa Alif Rahmawan2
Prafa Alif Rahmawan2 Mia Ratwita Andarsini3
Mia Ratwita Andarsini3 Indra Lesmana4
Indra Lesmana4 Emmanuel Jairaj Moses5
Emmanuel Jairaj Moses5 Nur Arzuar Abdul Rahim5
Nur Arzuar Abdul Rahim5 Narazah Mohd Yusoff1,5
Narazah Mohd Yusoff1,5Background: The frequency of the beta-thalassemia (β-thalassemia) gene in Indonesia ranges from 3 to 10%. However, in the East Java province, there is still limited information on the prevalence of β-thalassemia mutations in clinically diagnosed beta-thalassemia patients of East Java. Therefore, this study aimed to characterize β-thalassemia mutations in selected patients in the East Java province of Indonesia.
Methods: This is an analytical observational study. Diagnosis of β-thalassemia was based on clinical presentation, complete blood count (CBC), and hemoglobin (Hb) electrophoresis. Blood specimens taken from each patient in three ethylenediaminetetraacetic acid (EDTA) tubes were analyzed for CBC and Hb electrophoresis and processed for DNA extraction and subsequent polymerase chain reaction (PCR). Detection of mutations in Hemoglobin Subunit Beta (HBB) gene exons 1–3 of the β-thalassemia gene as the common mutation in Indonesia was done using PCR followed by Sanger sequencing.
Results: In total, 33 (n = 33) participants were involved in this study with ages ranging from 5 to 17 years comprising 19 women and 14 men. Their ethnic origins were Javanese (n = 30) and Chinese (n = 3). CBC results showed that mean ± standard deviation (SD) for Hb, red blood cell (RBC), mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), mean corpuscular hemoglobin concentration (MCHC), and red cell distribution width (RDW)-CV were 81.2 ± 7.0 g/L; 3.40 ± 0.39 × 109/L; 71.05 ± 5.72 fL; 24.12 ± 2.45 pg; 33.91 ± 1.47 g/dl; 24.38 ± 6.02%, respectively. Hb electrophoresis revealed that 5 out of 33 participants had beta-thalassemia and 28 out of 33 participants had hemoglobinopathy (Hb) E/beta-thalassemia. Results of Sanger sequencing showed the following genotype variations in the samples: 12 (36.4%) with βCD26/βIVS−I−5; 6 (18.2%) with βCD26/βCD35; 3 (9.1%) with βCD26/βIVS−I−2; 2 (6.1%) with βCD27/28/βCD40; 2 (6.1%) with βIVS−I−1/βCAP+1; and βCD26/βIVS−I−1; βIVS−I−5/βCAP+1; βIVS−I−5/βCD35; βCD26/βCD37; βCD26/βCD15; βCD26/βCD40; and βIVS−I−5/βCD19 in 1 (3%) sample, respectively, and 1 (3%) had no abnormality detected in sequencing even though electrophoresis showed abnormality in the migration pattern. The βCD26/βIVS−I−5 mutation was found in samples that were noted to have Hb E/beta-thalassemia on Hb electrophoresis.
Conclusion: The underlying genetic variations are heterogeneous in thalassemia patients in East Java, where 12 variants were found. The most common variant was βCD26/βIVS−I−5, which all accounted for Hb E/beta-thalassemia on Hb electrophoresis. Furthermore, 28 out of 33 participants had hemoglobinopathy (Hb) E/beta-thalassemia.
Beta thalassemia (β-thalassemia) is a disorder in hemoglobin synthesis characterized by decreased or absent β-globin chain synthesis. There are two (2) groups of β-thalassemia based on the amount of β-globin chain synthesis, namely, β0 if the globin chain is not synthesized at all, and β+ thalassemia if the globin chain synthesis is reduced. β0 thalassemia is mainly caused by point mutations in the coding region or exon-intron junction of the β-globin gene that causes premature stop codons or causes abnormal β-globin mRNA (1).
More than 200 different mutations underlying β-thalassemia have been identified (2, 3). These mutations are divided into β-globin gene deletions and non-deletional mutations that affect the transcription, processing, or translation of the globin messenger. Point mutations include mutations at the Catabolite Gene Activator Protein (CAP) site, frameshift, initiation site, nonsense mutation, polyA addition site mutation, promoter, and splicing mutation (2).
Some of these mutations underlie the different clinical manifestations in the affected patients and are classified as transfusion-dependent (TD) and non-transfusion-dependent (NTD) thalassemia, based on the transfusion needs of a patient (3). Patients classified as NTD thalassemia (NTDT) may not require frequent blood transfusions for survival, whereas those with TD thalassemia (TDT) require life-long regular blood transfusions (4).
The epidemiology of thalassemia involves more than 150 countries in the world that includes the Mediterranean, certain parts of North and West Africa, the Middle East, the Indian subcontinent, Southern Far East, with Southeast Asia having the highest prevalence (1, 4). Indonesia is not spared, having a high frequency of those with thalassemia genes. This is evident from epidemiological studies in Indonesia, which found that the frequency of beta-thalassemia genes ranged from 3 to 10% (5). Every year about 300,000–500,000 newborns are accompanied by severe hemoglobin abnormalities and 50,000–100,000 children die from thalassemia; 80% of them reside in developing countries. Data obtained from all teaching hospitals only registered about 7,670 thalassemia major patients throughout Indonesia. This number is still much lower than the actual estimated number. This could be because the types of gene mutations that exist in Indonesia vary from very severe to mild, thus they do not require transfusion (asymptomatic), resulting in under-diagnosis (5).
The molecular basis of the thalassemia's has been studied in many of the world's population and studies pertaining to the molecular basis of thalassemia in Indonesia have been reported (6, 7), especially in East Java. However, in these earlier reports, there were a limited number of samples, and studies were conducted about 10 years ago. A preliminary study conducted on 17 patients with TDT revealed the presence of seven (7) genotypic variations of beta-thalassemia (6). Currently, in Indonesia, the number of patients reported having severe thalassemia is increasing annually, and these numbers have increased four-fold in the last 20 years (7).
One of the reasons for this increase in the number of patients with thalassemia is due to high population migration to and from East Java over the last 10 years resulting in an increase in inter-ethnic marriages (8, 9).
Dr. Hospital Soetomo is a referral and the largest hospital in eastern Indonesia, with a land area of 166,061 hectares and 1,444 beds. This hospital was established in 1950 and provides services, education, and research functions, obtaining the Joint Commission International (JCI) accreditation in 2018 (10). Regarding data on new patients with TDT, there were 51 patients in 2014, 69 patients in 2015, 39 patients in 2016, 47 patients in 2017, 41 patients in 2018, and 57 patients in 2019. Routine pediatric outpatient visits were around 187 per month (unpublished data, Hematology-Oncology Division-Department of Pediatrics, Dr. Soetomo General Academic Hospital, 2022).
In East Java province, there is limited information on the prevalence of β-thalassemia mutations in the affected population. Therefore, this study aimed to characterize β-thalassemia mutations in clinically diagnosed patients with beta-thalassemia TDT in the East Java province of Indonesia.
The design of this study was analytical and observational. Participating patients were recruited from the One Day Care Unit of the Department of Pediatrics, Dr. Soetomo General Hospital, Surabaya, Indonesia. Previously diagnosed beta-thalassemia patients (<18 years old) who have undergone treatment that included blood transfusions at this institution were recruited. This study was carried out between July 2021 and January 2022.
Ethical clearance was obtained from the Ethics Committee of RSUD Dr. Soetomo No.: 0224/KEPK/VII/2021, and all of the patient's parents have agreed to give consent form.
Blood samples were taken from all patients on their follow-up prior to their blood transfusion. A total of 6 ml of blood was divided into two ethylenediaminetetraacetic acid (EDTA) tubes. The first tube was analyzed for complete blood count (CBC) using a Sysmex XN 1000 Analyzer (Sysmex Corporation, Kobe, Japan) and run for mini capillary hemoglobin electrophoresis (Sebia 9 Hydragel K20 Hemoglobin; Capillarys® Sebia, Lisses, France), while the other tube for DNA extraction using the QIAamp® DNA Blood Mini Kit lot no. kit. 166051764 (Qiagen GmbH, Hilden, Germany) according to the manufacturer's instructions. DNA samples were stored at −80°C, before subsequent analysis, i.e., performed polymerase chain reaction (PCR) and followed by the Sanger sequencing on these DNA samples. Ferritin, liver function tests, and renal function tests data were obtained from medical records based on the latest data. Molecular analysis was carried out at the Universitas Gajah Mada (UGM) Integrated Research and Testing Laboratory. Analytical statistics included the calculation of frequency distribution, mean, and standard deviation (SD).
Extracted DNA samples were subjected to Sanger sequencing. In this PCR reaction, the mixture contained 12.5 μl (Bioline My Taq HS Red Mix), 50–150 ng of genomic DNA, and two pairs of primers (Table 1) at concentrations of 0.4 μM each. The PCR reactions were performed using Bio-Rad T100 (Bio-Rad Laboratories, Inc., USA). The PCR cycle reactions were as follows: an initial 2 min of denaturation at 95°C; 15 s of denaturation (35 cycles) at 95°C; 15 s of annealing at 54°C; 15 s of extension at 72°C; and 2 min of extension at 72°C. Subsequently, 5 μl of the amplified product was aliquoted prior to visualization using gel electrophoresis. The gel preparation was as follows: 1% agarose gel in 1 × Tris-Borate-EDTA buffer. Electrophoresis was conducted for 25 min at 100 V and then viewed under a UV transilluminator prior to documentation (11).

Table 1. Sequence and size of the primers used for DNA amplification (17).
The PCR products encompassing the β globin genes were amplified with double reaction PCR. The amplified fragments were subsequently sequenced by Sanger methods by DNA Sequencing Services (UGM Integrated Research and Testing Laboratory). The sequences were analyzed and aligned using the Benchling web service (https://www.benchling.com/) with the reference sequence from NCBI Reference Seq: NG_059281 to determine the genotype (Applied Biosystems, 3500 Genetic Analyzer, Hitachi Corp Tokyo, Japan).
There were thirty-three study participants (19 women and 14 men) wherein 29 were Javanese and 4 were Chinese involved in this study with ages ranging from 5 to 17 years. CBC results showed that mean ± SD for Hb, red blood count (RBC), mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), mean corpuscular hemoglobin (MCHC), and red cell distribution width (RDW)-CV were 81.2 ± 7.0 g/L; 3.40 ± 0.39 × 109/L; 71.05 ± 5.72 fL; 24.12 ± 2.45 pg; 33.91 ± 1.47 g/dl; and 22.91 ± 8.66%, respectively. The patients have hypochromic microcytic anemia with anisocytosis on the peripheral blood film, the results of transaminase were mildly increased, however, the mean result of renal function was still within the normal range (Table 2).
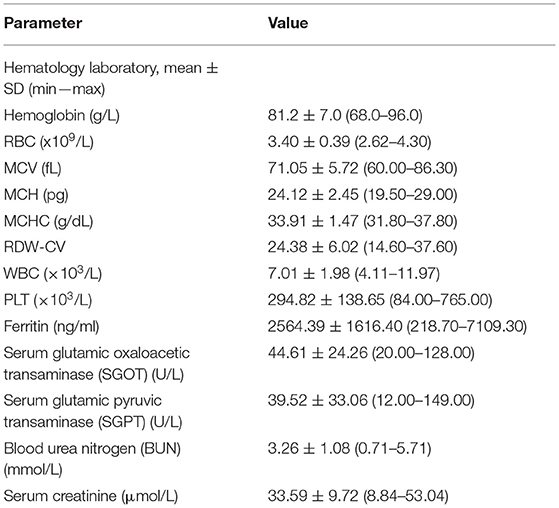
Table 2. Results of hematology, liver function test (LFT), ferritin, and renal function test (RFT) of all 33 patients.
Hemoglobin electrophoresis revealed that five (5) out of 33 patients had β-thalassemia and 28 out of 33 participants had hemoglobin variant (Hb) E/β-thalassemia. Results of Sanger sequencing showed the following genotype variations: 12 (36.4%) with βCD26/βIVS−I−5; 6 (18.2%) with βCD26/βCD35; 3 (9.1%) with βCD26/βIVS−I−2; 2 (6.1%) with βCD27/28/βCD40; 2 (6.1%) with βIVS−I−1/βCAP+1; and βCD26/βIVS−I−1; βIVS−I−5/βCAP+1; βIVS−I−5/βCD35; βCD26/βCD37; βCD26/βCD15; βCD26/βCD40; and βIVS−I−5/βCD19 in each sample (3%), respectively, while one (1) sample (3%) had no mutation detected even though the Hb electrophoresis results showed abnormality in the migration pattern.
There were 12 mutations detected, with βCD26/βIVS−I−5 being the most common identified, followed by βCD26/βCD35 and βCD26/βIVS−I−2, respectively (Table 3). All samples, which were identified to have the βCD26/βIVS−I−5 mutation, were noted to have Hb E/β-thalassemia on Hb electrophoresis. These samples were from 11 Javanese and 1 Chinese patient.
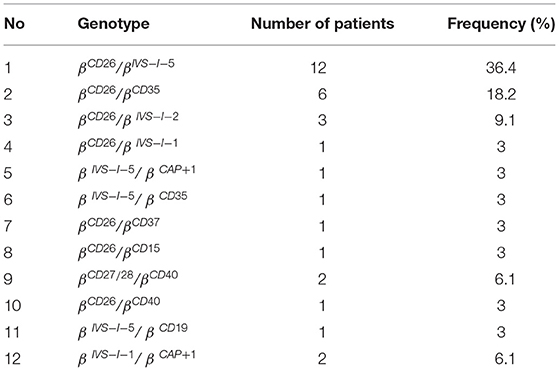
Table 3. The frequency of β-thalassemia genotype variations.
Patient nos. 17 and 18 were siblings and they had the same type of mutation but with different Hb electrophoresis results. Patient no. 18 who had HbE/β-thalassemia had lower Hb, MCV, MCH, and mean corpuscular hemoglobin concentration (MCHC) and higher RDW-CV as compared to patient no. 17 with β-thalassemia only, with results as follows: 81.0 vs. 94.0 g/L, 69.10 vs. 70.50 fL, 23.10 vs. 24.50 pg, 33.50 vs. 34.80 g/dl, 23.70 vs. 21.90, respectively (Table 4).
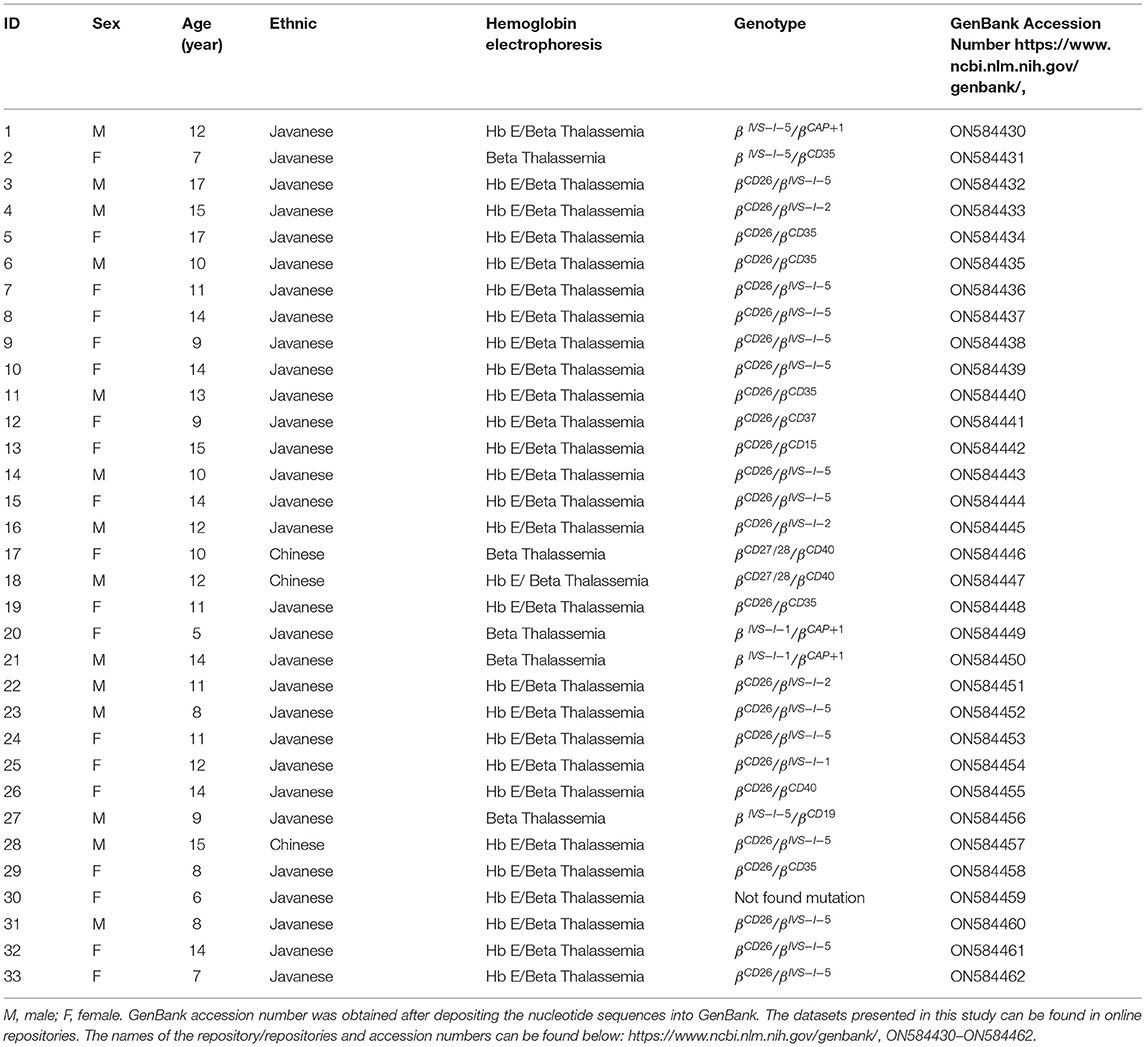
Table 4. Biodata, ethnicity groups, and results of hemoglobin electrophoresis, genotyping of β-thalassemia gene, and GenBank accession number in all 33 patients.
A total of 66 alleles were analyzed, 12 types of mutations from 64 alleles were found, while 2 alleles from one sample were not detected. Twelve types of mutations include mutations with frequency as follows: CD26 (G>A) 25 (40.63%), IVS-1-5 (G>C) 15 (23.44%), CD35 (delC) 7 (11%), IVS-1-2 (T>C); CAP+1 (A>C); CD40 (-G) 3 (4.7%), respectively, IVS-1-1 (G>T/A) 3 (4.7%); CD27/28 2 (3.1%), and CD37; CD15; and CD19 1 (1.6%), respectively.
We reported the genotype variation in 33 patients with TDT patients at Dr. Soetomo General Hospital, Surabaya, Indonesia. Hemoglobin Subunit Beta (HBB) genes were determined on exons 1–3 based on the most frequent abnormality in the Java population and had proven that 32 out of the 33 patients had their genotype successfully identified. The results showed that 12 genotype variations were found with the highest frequency found in the CD26/IVS-I-5 genotype (36.45%). This mutation was found in samples that have Hb E/beta-thalassemia on Hb electrophoresis and comprised 11 Javanese participants and one Chinese participant.
A previous study in East Java (6) showed a similar finding, which showed that 16 out of the 17 patients with TDT were compound heterozygotes for Hb E/beta-thalassemia (94.1%). The frequency calculated on the 34 independent alleles revealed Hb E 47.0%, IVS-I-5 20.6%; CD35 17.6%; CD15 (–T) 5.9%; CD41/42 2.9%; IVS-II-654 2.9%; and PolyA 2.9%.
The first report on beta-thalassemia genotype in Indonesia was by Lie-Injo (7). Out of 36 patients, 23 had β-thalassemia major and 13 had Hb E/beta-thalassemia with CD26 mutation (7) of which the results are comparable to our study.
In a previous study by Rujito et al. (12) regarding the genotype distribution of beta-thalassemia in the Javanese population of the south region of Central Java, in 209 patients, genotypes were as follows: CD26 /IVSI-5 (40.67%), followed by IVS-I-5/IVS-I-5 (14.83%). Of the 418 known alleles that were examined, IVS-I-5 was the most prevalent at 43.5% (12), which is comparable to our study.
The results of our study are also comparable to a recent study reported in Yogyakarta Special Region, conducted on 28 patients which showed six (6) β-globin gene mutations with IVS-1-5 (G>C) being the most dominant (71.4%) (13).
Another study in Bandung showed eight (8) types of mutations in 291 samples, with the highest prevalence of four (4) mutations, which were homozygous IVS1nt5 47.4%, heterozygote IVS1nt5/… (no paired mutation) 14.4%, heterozygous IVS1nt5/HbE 9.9%, and heterozygous IVS1nt5/IVS1nt1 5.4% (14). These results were slightly different from our study findings. This could be due to the different geographical locations of Java, which was West Java whereas our study was conducted in East Java.
A study conducted on 180 adolescent schoolgirls from East Java and West Java (15) found five (5) types of ß-globin gene polymorphism that were CD2, CD26/HbE, IVS1nt5, IVS2nt16, and IVS2nt74, which were quite similar in findings to our study; nevertheless CD2, IVS2nt16, and IVS2nt74 were not found in our cohort.
In another recent study on 31 beta-thalassemia patients from East Kalimantan, which predominantly consisted of Javanese (64.5%), the results revealed seven (7 types) mutant alleles, which were CD26/HbE at 48.4%, IVS-1-5 at 14.5%, IVS-1-2 at 12.9%, CD35 at 8.1%, and IVS-1-1 at 6.5%. These were partially comparable to our results especially CD26/HbE, IVS-1-2, and IVS-1-5 (16).
In comparison to other studies, our results revealed more mutations. It may reflect the variation of beta-globin mutation in the East Java population, since Dr. Soetomo Hospital is the biggest hospital in East Java and has become a referral hospital in East Java and eastern Indonesia. The results are fairly similar because West Java, Central Java, East Java, and the Special Region of Yogyakarta are all on one island, Java. Although East Kalimantan is part of Borneo island, i.e., a different island, the research was conducted in an area where the population is immigrants from Java, thus the results are almost the same.
Interestingly, 2 patients in our study were siblings (Table 4), and the CBC results in the sibling with HbE/β-thalassemia were much reduced than the sibling who had only β-thalassemia. This may be due to the fact that those with compound heterozygotes β-thalassemia/HbE have more severe clinical manifestations than those having only β-thalassemia. While one of them had no HbE detected in the Hb electrophoresis, both genotypes are identical, i.e., CD27/28/CD40. The explanation for this is that it may be related to the number of inherited CD27/28 alleles, where a small number of alleles will produce low HbE and cannot be detected on electrophoresis.
Noteworthily, there was no mutation detected in one of the patients via Sanger sequencing. Nevertheless, this patient was transfusion dependent thus indicating a severe form of thalassemia. This finding highlights the limitation of this study as it only targeted exons 1–3 of the HBB gene. It is also not feasible to sequence all 20 exons of this gene via Sanger sequencing. Therefore, this scenario creates a need for a more effective and high throughput technology in the form of Next-Generation Sequencing (NGS) technology, which would greatly aid in unraveling the entire spectrum of beta-thalassemia mutations in the Indonesian population. Furthermore, a larger cohort would give a more accurate picture of the underlying molecular spectrum of mutations in patients with TD and NTD beta-thalassemia in Indonesia.
The underlying genetic variations are heterogeneous in patients with TDT of East Java where there were 12 variants found, the most common of which is βCD26/βIVS−I−5, found in all patients with Hb E/beta-thalassemia detected via Hb electrophoresis.
The results of this study provide data that may be useful for the prevention and control strategies of thalassemia in Indonesia.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession numbers can be found below: https://www.ncbi.nlm.nih.gov/genbank/: ON584430, ON584431, ON584432, ON584433, ON584434, ON584435, ON584436, ON584437, ON584438, ON584439, ON584440, ON584441, ON584442, ON584443, ON584444, ON584445, ON584446, ON584447, ON584448, ON584449, ON584450, ON584451, ON584452, ON584453, ON584454, ON584455, ON584456, ON584457, ON584458, ON584459, ON584460, ON584461, ON584462.
The studies involving human participants were reviewed and approved by Ethics Committee of RSUD Dr. Soetomo with No.: 0224/KEPK/VII/2021. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
YH designed and wrote the manuscript. YS collected samples and processed until DNA extraction, then sent to UGM. YI assisted in handling Hb electrophoresis, complete blood count. PR wrote the proposal to obtain funding. MA arrange for recruitment of pediatric patients. NY proofread and assisted with corrections until final version of manuscript. All authors contributed to the article and approved the submitted version.
The authors thank the Faculty of Medicine Universitas Airlangga for funding the research, granted by Decree of the Chancellor of Airlangga University Number 212/UN3/2021.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor ZAL declared a past co-authorship with the author NY.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank all the patients who participated and the hospital staffs who have rendered their assistance and contributions to this study.
1. Tatu T. Laboratory Diagnosis of β-Thalassemia and HbE. Vienna: IntechOpen (2020). doi: 10.5772/intechopen.90317
2. Kaushansky K LM, Prchal Josef LMM, Press Oliver BL, Caligiuri M. Williams Hematology 9th edition. New York: McGraw-Hill Education/Medical (2015).
3. Bajwa H, Basit H. Thalassemia. Orlando, FL: StatPearls Publishing LLC (2022). p. 1–38. Available online at: https://www.ncbi.nlm.nih.gov/books/NBK545151/?report=reader#_NBK545151_pubdet_ (accessed March 8, 2022).
4. Sleiman J, Tarhini A, Bou-Fakhredin R, Saliba AN, Cappellini MD, Taher AT. Non-transfusion-dependent thalassemia: an update on complications and management. Int J Mol Sci. (2018) 19:10182. doi: 10.3390/ijms19010182
5. Minister of Health of the Republic of Indonesia. Pedoman Nasional Pelayanan Kedokteran Tata laksana Thalasemia. Indonesia: MHRI (2018).
6. Hernanda PY, Tursilowati L, Arkesteijn SGJ, Ugrasena IDG, Larasati MCS, Soeatmadji SM, et al. Towards a prevention program for β-thalassemia. The molecular spectrum in East Java, Indonesia. Hemoglobin. (2012) 36:1–6. doi: 10.3109/03630269.2011.642914
7. Lie-Injo LE, Cai SP, Wahidijat I, Moeslichan S, Lim ML, Evangelista L, et al. Beta-thalassemia mutations in Indonesia and their linkage to beta haplotypes. Am J Human Genetic. (1989) 45:971–5.
8. Setyaningtyas FY, Pratomo DS, Syafitri W. An analysis of factors affecting recent out-migration in East Java. South East Asia J Contemp Bus Econ Law. (2021) 24:169–74.
9. Yulianto JE, Hodgetts D, King P, Liu JH. Navigating tensions in inter-ethnic marriages in Indonesia: cultural, relational, spatial and material considerations. Int J Intercult Relat. (2022) 86:227–39. doi: 10.1016/j.ijintrel.2021.12.008
10. Edwar PPM, Krisna R, Murniati T, Mustakim Z, Mustikarani R, Amanda YD, et al. Profil dan Panduan Informasi RS Pendidikan RSUD Dr. Soetomo. Revision ed. Surabaya: RSUD Dr Soetomo-FKUA (2019). p. 1–11.
11. Kountouris P, Lederer CW, Fanis P, Feleki X, Old J, Kleanthous M. IthaGenes: an interactive database for hemoglobin variations and epidemiology. PLoS ONE. (2014) 9:103020. doi: 10.1371/journal.pone.0103020
12. Rujito L, Basalamah M, Mulatsih S, Sofro ASM. Molecular scanning of β-thalassemia in the southern region of Central Java, Indonesia; a step towards a local prevention program. Hemoglobin. (2015) 39:330–3. doi: 10.3109/03630269.2015.1065420
13. Handayani NSN, Husna N, Rahmil G, Ghifari RA, Widyawati L, Lesmana I. Splice-site and frameshift mutations of β-globin gene found in thalassemia carrier screening in Yogyakarta Special Region, Indonesia. Indonesian Biomed J. (2020) 13:55–60. doi: 10.18585/inabj.v13i1.1406
14. Maskoen AM, Rahayu NS, Reniarti L, Susanah S, Laksono B, Fauziah PN, et al. Mutation spectrum of Beta-globin gene in thalassemia patients at Hasan Sadikin Hospital-West Java Indonesia. Cell Mol Biol (Noisy-Le-Grand). (2017) 63:22–4. doi: 10.14715/cmb/2017.63.12.6
15. Adhiyanto C, Susianti Y, Rahmawati NM, Badriah F, Harriati ZR, et al. Screening of Beta-globin gene mutations in adolescent schoolgirls in rural Malang and Sukabumi city, Java Province, Indonesia. Adv Health Sci Res. (2017) 10:183–6. doi: 10.2991/ichlas-17.2017.47
16. Susanto ZA, Siswandari W, Rujito L. Cd60 (GTG > GAG)/Hb Cagliari mutation was found in scanning of β-thalassemia alleles from patients of East Kalimantan, Indonesia. Mol Genet Metab Rep. (2020) 22:1–3. doi: 10.1016/j.ymgmr.2019.100550
Keywords: β-thalassemia, β-thalassemia mutations, East Java, Indonesia, anemia
Citation: Hernaningsih Y, Syafitri Y, Indrasari YN, Rahmawan PA, Andarsini MR, Lesmana I, Moses EJ, Abdul Rahim NA and Yusoff NM (2022) Analysis of Common Beta-Thalassemia (β-Thalassemia) Mutations in East Java, Indonesia. Front. Pediatr. 10:925599. doi: 10.3389/fped.2022.925599
Received: 21 April 2022; Accepted: 20 June 2022;
Published: 15 July 2022.
Edited by:
Zarina Abdul Latiff, National University of Malaysia, MalaysiaReviewed by:
Catherine Lynn T. Silao, University of the Philippines Manila, PhilippinesCopyright © 2022 Hernaningsih, Syafitri, Indrasari, Rahmawan, Andarsini, Lesmana, Moses, Abdul Rahim and Yusoff. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yetti Hernaningsih, eWV0dGktaEBmay51bmFpci5hYy5pZA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.