
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pediatr., 27 June 2022
Sec. Pediatric Gastroenterology, Hepatology and Nutrition
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.914790
This article is part of the Research TopicInsights in Pediatric Pancreatology 2022View all 12 articles
 Isabelle R. McKay1
Isabelle R. McKay1 Chee Y. Ooi2,3*
Chee Y. Ooi2,3*Cystic fibrosis (CF) is a common disorder of autosomal recessive inheritance, that once conferred a life expectancy of only a few months. Over recent years, significant advances have been made to CF therapeutic approaches, changing the face of the disease, and facilitating the partial restoration of pancreatic function. This mini review summarizes the current landscape of exocrine pancreatic management in CF and explores areas for future direction and development.
Cystic fibrosis (CF) is one of the most common recessive genetic disorders worldwide, with wide-ranging health implications. While CF is characterized as an illness of pulmonary morbidity and mortality, it is a multisystem disorder encompassing sequelae in the gastrointestinal, hepatobiliary, and pancreatic systems (1). CF occurs due to mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Over 2,000 mutations, with varying functional consequences, have been identified to date, resulting in a spectrum of disease phenotypes. The CF gene encodes the CFTR protein responsible for driving chloride, bicarbonate and fluid secretion in affected epithelial surfaces. Dysfunction in the CFTR protein results in thick, inspissated secretions which in turn leads to obstruction, infection, inflammation and ultimately destruction of affected organs (2). These processes result in clinical manifestations such as chronic sinopulmonary diseases, exocrine and endocrine pancreatic diseases, intestinal obstruction, cirrhosis and obstructive azoospermia from atrophic or absent vasa deferens (3). This mini-review will focus on the exocrine pancreatic disease in CF, and how presentation and management has evolved with the introduction of CFTR-modulating therapies.
Pancreatic dysfunction in CF is a result of ductal obstruction from early in life. The specific mechanisms behind this process are multifactorial and incompletely understood, but are thought to hinge on dysregulated bicarbonate buffering, altered chloride flux and the production of pro-inflammatory pancreatic secretions. Our understanding of the pathophysiology has evolved exponentially in recent years, and is continuing to expand rapidly. The first pathological descriptions of “fibrocystic disease” of the pancreas were published by Dr. Dorothy Andersen in 1938 (4), but it was not until 1989, almost four decades later, that the CFTR gene was first recognized by Riordan and colleagues, demonstrating the clear genetic basis for the multisystem phenotype of CF (5).
In the 30 years since, our understanding of the role of CFTR in pancreatic function has deepened, guiding CF management beyond a “blanket” approach and toward more directed therapy. Today, CFTR has been established as a key modulator of chloride and bicarbonate transport, with downstream implications for several organ systems. In the small pancreatic ducts, this ionic flux is crucial in the production of alkaline fluid, the neutralization of gastric acid and the maintenance of a functional environment for digestive enzymes (2).
In the pancreatic ductal epithelia, defective CFTR results in ductal secretions with a lower pH (secondary to reduced bicarbonate buffering), a lower volume (secondary to reduced sodium chloride-driven osmosis) and an increased viscosity (secondary to protein hyperconcentration) (6, 7). This process begins in utero, resulting in early acinar plugs consisting mainly of zymogen material. As exocrine atrophy progresses, the ability of the pancreas to produce zymogens decreases, and these plugs come to contain a higher proportion of mucins secondary to ductal metaplasia (8). Ultimately, the end effects of CFTR dysregulation have been widely observed to result in pancreatic ductal obstruction and zymogen accumulation, leading to fibroinflammatory changes and parenchymal injury (2, 9).
The spectrum of phenotypic presentation of exocrine pancreatic disease in CF closely correlates with individual genotype. While >2,000 mutations have been identified to date, with more expected to be uncovered with time, they can be broadly categorized into six classes in relation to the degree of CFTR protein production and function. These classes are summarized in Table 1 below.
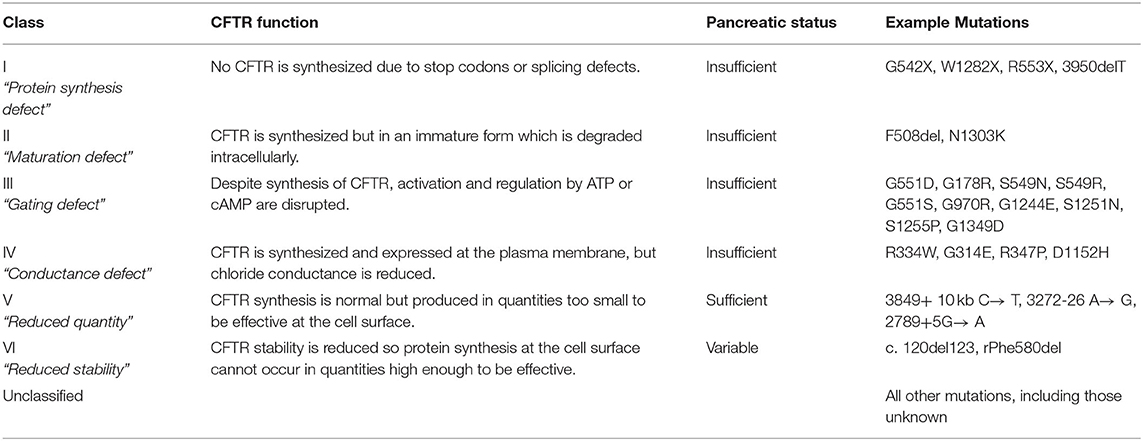
Table 1. An overview of the genotype classes in CF, with their associated pancreatic phenotype (11–13).
Genotype classes I, II and III are commonly associated with pancreatic insufficiency, due to a greater degree of CFTR deficiency and dysfunction. Genotype classes IV, V, and VI or compound heterozygous individuals with one less severe allele, tend to retain some level of exocrine function as CFTR is still produced to some extent (Table 1). Response to therapeutic interventions varies between genotype classes, and varies further within each class itself. While genotype provides one method of classifying CF disease to guide investigation and management, it is important to recognize that outcomes in CF are also influenced by a number of other determinants including epigenetic factors, genetic modifiers, environmental factors and socioeconomic status (11).
The identification of CFTR as the genetic basis for CF disease has turned scientific research toward precision medicine, and the early 2000s saw the introduction of CFTR modulator drugs. CFTR modulators are designed to support or restore the functionality of CFTR, and are classified into five key groups based on mechanism: potentiators, correctors, stabilizers, read-through agents, and amplifiers (3). Currently, only four agents have been made available on the pharmaceutical market, and are approved for use only in specific genotypes.
Potentiators restore or augment the cAMP mediated CFTR gating, allowing for some degree of CFTR-dependent transport to occur. Approximately 5% of CF mutations are a gating or conductance deficit, and it is this proportion of the CF population that will benefit from potentiator therapy. These mutations tend to fall within Classes III and IV (Table 1). Currently, the only available potentiator is ivacaftor, which is approved use either as a monotherapy, or as combination therapy with the correctors tezacaftor or lumacaftor. While the mechanism of action is yet to be fully elucidated, clinical outcomes demonstrate significant improvement amongst eligible patients commenced on therapy, ranging from decreased frequency of pulmonary exacerbations, to nutritional improvement and rescued pancreatic function (14, 15). While there are several other potentiators currently under clinical evaluation, ivacaftor remains the only potentiator available for patient use.
Correctors (e.g., tezacaftor, lumacaftor, and elexacaftor) assist CFTR protein structuring and trafficking. While the specifics of activity vary between individual agents, the mechanism of the corrector class is considered to be either direct (binding to the misfolded protein itself and “chaperoning” it through the endoplasmic reticulum) or indirect (through proteostasis regulation) (3). Mistrafficking is the most common mutation type in CF, notably including the F508del mutation.
Stabilizing therapies target Class VI mutations, wherein the CFTR protein is present at the plasma membrane but has reduced availability due to increased lysosomal degradation. Stabilizers function to anchor the protein at the cell surface, preventing premature removal and destruction. While lumacaftor has been shown to transiently increase CFTR stability, ongoing investigation into agents with longer-term benefits are ongoing (16).
One of the more severe phenotypes in CF results from defective CFTR synthesis in the first instance, usually due to the introduction of a premature termination codon (PTC) into the protein mRNA (Class I mutations). Read-through agents allow the protein translation process to “skip over” the PTC through recruiting an alternative amino acid in its place, facilitating the production of a full-length protein (17). Aminoglycosides such as gentamicin have demonstrated read-through capabilities in early experimental studies, but the practical application of these properties is limited by the toxicity profile seen with longer-term dosing. While some trials are currently examining aminoglycoside derivatives with stronger read-through capability and less toxicity, nothing has been approved for use in CF to date (18, 19).
Class V mutations encompass those genotypes resulting in reduced synthesis or maturation of the CFTR protein. Amplifiers target this class of mutation, increasing the expression of CFTR mRNA and the downstream protein production load (20). Nesolicaftor is currently in the midst of phase 3 trials (Proteostasis Therapeutics), having shown promising results throughout earlier studies with significantly higher CFTR mRNA levels when used in combination with existing corrector and potentiator therapies (21, 22).
Two key clinical manifestations present themselves as hallmarks of exocrine pancreatic disease in CF: (1) pancreatic insufficiency, and (2) symptomatic pancreatitis among a subset of people with pancreatic sufficient (PS) CF. Each of these confer a distinct disease burden and occur within a specific subset of the CF population, shaping presentation and management.
Approximately 85% of people with CF are pancreatic insufficient (PI) from early in life (23). A near absolute loss in exocrine output results in a disease of maldigestion, characterized by steatorrhoea, failure to thrive, and fat-soluble vitamin deficiencies. Left unrecognized, this population historically died in early infancy from malnutrition, before the now-familiar pulmonary manifestations of CF even took root (24). Under- or malnutrition (as measured by BMI) has been shown in epidemiological studies to be closely linked with poor pulmonary and survival outcomes in CF (25). Nowadays, the appropriate and timely initiation of pancreatic enzyme replacement therapy (PERT), coupled with a high energy diet, allows these individuals to facilitate nutritional digestion and maintain adequate growth and development; hence early recognition and management are essential. Even with PERT treatment, the clinical consequences of PI are ongoing and continues to represent a large proportion of CF morbidity and mortality, leading to malnutrition, poor weight gain and a decreased ability to withstand intercurrent clinical insults (26).
The PI status should always be confirmed on testing. Fecal elastase is the most commonly utilized test in clinical practice for assessing exocrine pancreatic function. Traditionally, a fecal elastase cutoff of <200 μg/g as indicative of PI is used although a lower cutoff of 100 μg/g has been reported to be of greater predictive value for ruling out PI and minimize false positive results due to dilution of feces caused by non-pancreatic intestinal causes (e.g., short gut syndrome) (27). Other indirect measures of exocrine pancreatic function include fecal chymotrypsin and serum trypsinogen. These latter two tests have a lower sensitivity and specificity than fecal elastase, and their use in the diagnostic setting is curbed by some key clinical limitations. Fecal chymotrypsin is a commercially available enzyme, so it cannot be reliably measured in patients prescribed PERT (28). Serum trypsinogen is not specific for exocrine pancreatic function, and can be elevated in other states of disease including acute pancreatitis and is not widely available (29). Direct tests of pancreatic function, such as secretin or cholecystokinin testing (using a dreiling tube or endoscope), are more sensitive and specific, but less commonly used due to their technical nature and poor patient tolerance and/or need for general anesthesia (in children) (30).
Approximately 15% of the CF cohort are PS, born with at least 2% of residual pancreatic reserve, enough to adequately digest and absorb nutrients (26). While PS individuals tend to exhibit a less severe phenotype overall, it is in this population that symptomatic pancreatitis may occur, at a proportion of ~20%. It is thought to result from an altered ratio of acinar reserve and ductal obstruction, as conceptualized in Figure 1. Compounding this process, impaired bicarbonate secretion leads to altered luminal pH, promoting ongoing acute tissue inflammation and the perpetuation of pancreatitis in the chronic period (31). This pathology relies on a degree of acinar function to be preserved in the presence of significant ductal obstruction, hence a negligible prevalence of pancreatitis amongst the PI group.
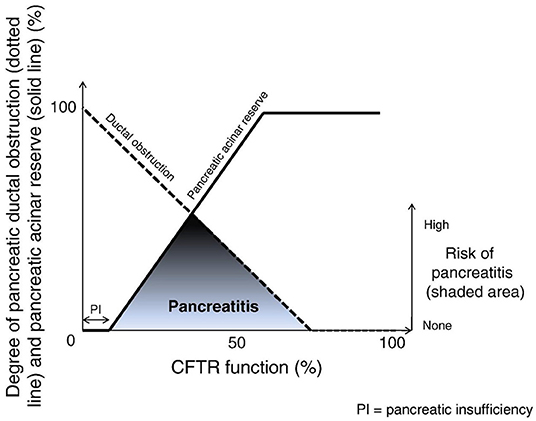
Figure 1. The development of pancreatitis in the context of CFTR-related contributors. Adapted from Ooi et al. (10).
As well as genotype being a key determinant, risk of pancreatitis is also amplified in the context of cigarette or alcohol use, both of which independently decrease CFTR function (32, 33). Pancreatitis in the CF cohort is diagnosed with classical criteria of archetypal symptomatology, serum amylase/lipase greater than three times the upper limit of normal, and/or consistent findings on imaging (34).
It is also worth noting that in the context of the progressive nature of pancreatic disease in CF, a percentage of PS individuals will become PI later in life. The likelihood of this level of pancreatic destruction is largely informed by genotype (e.g., carriage of classes I, II or III mutations on both alleles) and previous bouts of symptomatic pancreatitis (10). This suggests that recurrent attacks of symptomatic pancreatitis heralds progressive deterioration in exocrine function, underscoring the importance of serially monitoring function amongst the PS group.
CFTR modulator treatment eligibility and planning is generally based on genotype classing (as described in Table 1). However, this patient classification system does not incorporate likely pancreatic response into the decision-making process. As CFTR modulator use has increased, longitudinal evidence comes forth, describing three different patient groups with distinct pancreatic presentations and response to therapy.
Pancreatic dysfunction in CF begins in utero, and while PERT prescription can assist in mitigating some of the clinical ramifications, exocrine disease persists throughout life (35). As CFTR modulators have become established as a key tenet of CF management, their wide-ranging effects on health beyond pulmonary capacity have been brought to the fore. Emerging evidence has demonstrated improvement and recovery in exocrine pancreatic function, with implication for growth and nutritional outcomes later in life. The ARRIVAL trial noted improvements in measured pancreatic markers such as fecal elastase (FE) and immunoreactive trypsinogen (IRT) when ivacaftor was introduced in young children between 12 and 24 months (36), and the KIWI/KLIMB studies demonstrated similar improvements amongst a group in early childhood (aged 2–5 years) (37, 38). However, the magnitude of these benefits is curbed as the introduction age of ivacaftor increases, and ferret models have demonstrated that withdrawal of therapy reinstates exocrine disease (39, 40). Ultimately, these findings support the concept that the “window of opportunity” to rescue exocrine pancreatic function in CF occurs early in life, hinging on early and sustained modulator therapy.
The “window of opportunity” in initiating CFTR modulator treatment is likely to be from as early in life as possible. However, a recent ivacaftor study in ferret models demonstrated partial protection from disease progression when the therapy was commenced in utero. Ferrets in the treatment group demonstrated similar growth rates as wild-type animals, and continued on a normal growth trajectory while nursing even without PERT treatment (40). Corroborating this proof-of-concept, a recent human case report provides details of a child born to a F508del homozygous mother on elexacaftor/tezacaftor/ivacaftor treatment. Despite inheriting a F508del homozygous genotype themselves, this child did not meet the laboratory criteria for the IRT CF newborn screening, and was born PS with growth tracking along the 85th centile (41). Together, these studies support the finding that pancreatic disease begins in utero, and raises the possibility that modulator treatment commenced even prior to birth may slow or perhaps prevent exocrine disease from taking hold. Currently, no modulator therapy is approved for use earlier than 2 years of age.
Rescuing exocrine pancreatic function in CF has clear benefits for nutrition and growth. However, increasing pancreatic reserve in CF poses its own risks, as a growing body of evidence highlights links to the development of symptomatic acute pancreatitis amongst a subset of patients who were PI prior to commencement of modulators, corroborated by the findings of recent case reports. Gould et al. describe a series of five patients, all PI, who developed a classical presentation of acute pancreatitis at a median of 30 months following commencement of modulator therapy. Of these five, three had regained some level of exocrine function with FE measurements above 100 μg/g (42). Megalaa et al. describe a similar case report of a 10 year old child, realized to have regained PS status only after an episode of acute pancreatitis (43). In keeping with the model conceptualized by Ooi and colleagues in 2011 (Figure 1), this suggests that while CFTR modulators may effectively increase pancreatic acinar reserve, in the setting of ongoing ductal obstruction, this can result in a heightened risk of pancreatitis (10). This speaks to the risk profile of CFTR modulators, highlighting that their health benefits must be considered in the context of potential serious complications which clinicians must remain vigilant for amongst this population.
Conversely, PS patients commenced on modulator treatment appear to accelerate beyond this critical ratio of acinar reserve and ductal obstruction (Figure 1), with a subsequent decline in the prevalence of pancreatitis amongst this group. Akshintala et al. retrospectively reviewed a small cohort of adult CF patients with a history of pancreatitis in the preceding 2 years, highlighting that none of these 15 individuals developed pancreatitis during their follow up period (mean 36 months) (44). Ramsey and colleagues supported these findings 3 years onwards, demonstrating a significant reduction in pancreatitis-related hospitalizations amongst those commenced on CFTR modulators amongst both PI and PS patients, with a greater relative risk reduction within the PS group (45). Overall, these findings suggest that in those suffering from recurrent pancreatitis, or who have a pre-existing risk of pancreatitis, CFTR modulators may assist in shifting away from this “risk window,” alleviating ductal obstruction enough to improve pancreatic output without inducing further inflammation.
Pancreatic disease represents a significant proportion of CF-related morbidity and mortality. Where treatment previously focused mitigating the effects of downstream sequalae, the research landscape has shifted now to focus on addressing the central CFTR mutation at the root. As CFTR modulators become established as a cornerstone of CF management, the limitations of these novel agents are brought to the fore. The risk-benefit profile of these therapies varies for three CF cohort subsets, depending on pre-existing pancreatic function and risk of pancreatitis. This classification of modulator-eligible patients encourages a patient-centered treatment approach, where a distinct risk monitoring process may facilitate the early recognition of key complications unique to each population. The temporal outcomes of CFTR modulator use in the context of longer-term pancreatic complications including CF-related diabetes and malignancy are yet to be established, but with demonstrable benefits in the short-term setting, positive effects may be anticipated.
IM and CO conceptualized the topic and drafted review design. IM analyzed the literature and drafted the manuscript. CO refined the manuscript and provided guidance on literature interpretation. All authors made significant and direct contributions to the work and approve it for publication.
CO has served as a consultant and received research funding (unrelated to the content of this manuscript) from Vertex Pharmaceuticals.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Donaldson SH, Boucher RC. Pathophysiology of cystic fibrosis. Annales Nestlé. (2006) 64:101–9. doi: 10.1159/000095374
2. Gibson-Corley KN, Meyerholz DK, Engelhardt JF. Pancreatic pathophysiology in cystic fibrosis. J Pathol. (2016) 238:311–20. doi: 10.1002/path.4634
3. Lopes-Pacheco M. CFTR modulators: shedding light on precision medicine for cystic fibrosis. Front Pharmacol. (2016) 7:275. doi: 10.3389/fphar.2016.00275
4. Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathologic study. Am J Dis Child. (1938) 56:344–99. doi: 10.1001/archpedi.1938.01980140114013
5. Riordan JR, Rommens JM, Kerem BS, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. (1989) 245:1066–73. doi: 10.1126/science.2475911
6. Kopelman H, Corey M, Gaskin K, Durie P, Weizman Z, Forstner G. Impaired chloride secretion, as well as bicarbonate secretion, underlies the fluid secretory defect in the cystic fibrosis pancreas. Gastroenterology. (1988) 95:349–55. doi: 10.1016/0016-5085(88)90490-8
7. Kopelman H, Durie P, Gaskin K, Weizman Z, Forstner G. Pancreatic fluid secretion and protein hyperconcentration in cystic fibrosis. N Engl J Med. (1985) 312:329–34. doi: 10.1056/NEJM198502073120601
8. Angyal D, Bijvelds MJC, Bruno MJ, Peppelenbosch MP, de Jonge HR. Bicarbonate Transport in cystic fibrosis and pancreatitis. Cells. (2021) 11:54. doi: 10.3390/cells11010054
9. Wilschanski M, Durie PR. Patterns of GI disease in adulthood associated with mutations in the CFTR gene. Gut. (2007) 56:1153–63. doi: 10.1136/gut.2004.062786
10. Ooi CY, Dorfman R, Cipolli M, Gonska T, Castellani C, Keenan K, et al. Type of CFTR mutation determines risk of pancreatitis in patients with cystic fibrosis. Gastroenterology. (2011) 140:153–61. doi: 10.1053/j.gastro.2010.09.046
11. Lopes-Pacheco M. CFTR modulators: the changing face of cystic fibrosis in the era of precision medicine. Front Pharmacol. (2020) 10:1662. doi: 10.3389/fphar.2019.01662
12. Durda-Masny M, Gozdzik-Spychalska J, John A, Czaiński W, Strózewska W, Pawłowska N, et al. The determinants of survival among adults with cystic fibrosis—a cohort study. J Physiol Anthropol. (2021) 40:19. doi: 10.1186/s40101-021-00269-7
13. Marson FAL, Bertuzzo CS, Ribeiro JD. Classification of CFTR mutation classes. Lancet Respir Med. (2016) 4:e37–8. doi: 10.1016/S2213-2600(16)30188-6
14. McKone EF, Borowitz D, Drevinek P, Griese M, Konstan MW, Wainwright C, et al. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-CFTR mutation: a phase 3, open-label extension study (PERSIST). Lancet Respir Med. (2014) 2:902–10. doi: 10.1016/S2213-2600(14)70218-8
15. Nichols A, Davies J, Jones D, Carr S. Restoration of exocrine pancreatic function in older children with cystic fibrosis on ivacaftor. Paediatr Respir Rev. (2020) 35:99–102. doi: 10.1016/j.prrv.2020.04.003
16. He L, Kota P, Aleksandrov AA, Cui L, Jensen T, Dokholyan NV, et al. Correctors of ΔF508 CFTR restore global conformational maturation without thermally stabilizing the mutant protein. FASEB J. (2013) 27:536–45. doi: 10.1096/fj.12-216119
17. Pranke I, Bidou L, Martin N, Blanchet S, Hatton A, Karri S, et al. Factors influencing readthrough therapy for frequent cystic fibrosis premature termination codons. ERJ Open Res. (2018) 4:00080-2017. doi: 10.1183/23120541.00080-2017
18. Leubitz A, Frydman-Marom A, Sharpe N, van Duzer J, Campbell KC, Vanhoutte F. Safety, tolerability, and pharmacokinetics of single ascending doses of ELX-02, a potential treatment for genetic disorders caused by nonsense mutations, in healthy volunteers. Clin Pharmacol Drug Dev. (2019) 8:984–94. doi: 10.1002/cpdd.647
19. Sharma J, Keeling KM, Rowe SM. Pharmacological approaches for targeting cystic fibrosis nonsense mutations. Eur J Med Chem. (2020) 200:112436. doi: 10.1016/j.ejmech.2020.112436
20. Giuliano KA, Wachi S, Drew L, Dukovski D, Green O, Bastos C, et al. Use of a high-throughput phenotypic screening strategy to identify amplifiers, a novel pharmacological class of small molecules that exhibit functional synergy with potentiators and correctors. SLAS Discov. (2018) 23:111–21. doi: 10.1177/2472555217729790
21. Gramegna A, Contarini M, Aliberti S, Casciaro R, Blasi F, Castellani C. From ivacaftor to triple combination: a systematic review of efficacy and safety of CFTR modulators in people with cystic fibrosis. Int J Mol Sci. (2020) 21:5882. doi: 10.3390/ijms21165882
22. Molinski SV, Ahmadi S, Ip W, Ouyang H, Villella A, Miller JP, et al. Orkambi® and amplifier co-therapy improves function from a rare CFTR mutation in gene-edited cells and patient tissue. EMBO Mol Med. (2017) 9:1224–43. doi: 10.15252/emmm.201607137
23. Stallings VA, Stark LJ, Robinson KA, Feranchak AP, Quinton H. Evidence-based practice recommendations for nutrition-related management of children and adults with cystic fibrosis and pancreatic insufficiency: results of a systematic review. J Am Diet Assoc. (2008) 108:832–9. doi: 10.1016/j.jada.2008.02.020
24. Reid DW, Blizzard CL, Shugg DM, Flowers C, Cash C, Greville HM. Changes in cystic fibrosis mortality in Australia, 1979–2005. Med J Australia. (2011) 195:392–5. doi: 10.5694/mja10.11229
25. Lai H-C, Corey M, FitzSimmons S, Kosorok MR, Farrell PM. Comparison of growth status of patients with cystic fibrosis between the United States and Canada. Am J Clin Nutr. (1999) 69:531–8. doi: 10.1093/ajcn/69.3.531
26. Singh VK, Schwarzenberg SJ. Pancreatic insufficiency in cystic fibrosis. J Cystic Fibrosis. (2017) 16:S70–8. doi: 10.1016/j.jcf.2017.06.011
27. Beharry S, Ellis L, Corey M, Marcon M, Durie P. How useful is fecal pancreatic elastase 1 as a marker of exocrine pancreatic disease? J Pediatr. (2002) 141:84–90. doi: 10.1067/mpd.2002.124829
28. Niederau C, Grendell JH. Diagnosis of chronic pancreatitis. Gastroenterology. (1985) 88:1973–95. doi: 10.1016/0016-5085(85)90029-0
29. Couper RT, Corey M, Durie PR, Forstner GG, Moore DJ. Longitudinal evaluation of serum trypsinogen measurement in pancreatic-insufficient and pancreatic-sufficient patients with cystic fibrosis. J Pediatr. (1995) 127:408–13. doi: 10.1016/S0022-3476(95)70072-2
30. Lankisch PG, Schreiber A, Otto J. Pancreolauryl test. Evaluation of a tubeless pancreatic function test in comparison with other indirect and direct tests for exocrine pancreatic function. Dig Dis Sci. (1983) 28:490–3. doi: 10.1007/BF01308149
31. Behrendorff N, Floetenmeyer M, Schwiening C, Thorn P. Protons released during pancreatic acinar cell secretion acidify the lumen and contribute to pancreatitis in mice. Gastroenterology. (2010) 139:1711–20. e5. doi: 10.1053/j.gastro.2010.07.051
32. Raju SV, Jackson PL, Courville CA, McNicholas CM, Sloane PA, Sabbatini G, et al. Cigarette smoke induces systemic defects in cystic fibrosis transmembrane conductance regulator function. Am J Respir Crit Care Med. (2013) 188:1321–30. doi: 10.1164/rccm.201304-0733OC
33. Maléth J, Balázs A, Pallagi P, Balla Z, Kui B, Katona M, et al. Alcohol disrupts levels and function of the cystic fibrosis transmembrane conductance regulator to promote development of pancreatitis. Gastroenterology. (2015) 148:427–39.e16. doi: 10.1053/j.gastro.2014.11.002
34. Morinville VD, Husain SZ, Bai H, Barth B, Alhosh R, Durie PR, et al. Definitions of pediatric pancreatitis and survey of present clinical practices. J Pediatr Gastroenterol Nutr. (2012) 55:261–5. doi: 10.1097/MPG.0b013e31824f1516
35. Bass R, Brownell JN, Stallings VA. The impact of highly effective CFTR modulators on growth and nutrition status. Nutrients. (2021) 13:2907. doi: 10.3390/nu13092907
36. Rosenfeld M, Wainwright CE, Higgins M, Wang LT, McKee C, Campbell D, et al. Ivacaftor treatment of cystic fibrosis in children aged 12 to 24 months and with a CFTR gating mutation (ARRIVAL): a phase 3 single-arm study. Lancet Respir Med. (2018) 6:545–53. doi: 10.1016/S2213-2600(18)30202-9
37. Rosenfeld M, Cunningham S, Harris WT, Lapey A, Regelmann WE, Sawicki GS, et al. An open-label extension study of ivacaftor in children with CF and a CFTR gating mutation initiating treatment at age 2-5 years (KLIMB). J Cyst Fibros. (2019) 18:838–43. doi: 10.1016/j.jcf.2019.03.009
38. Davies JC, Cunningham S, Harris WT, Lapey A, Regelmann WE, Sawicki GS, et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2-5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med. (2016) 4:107–15. doi: 10.1016/S2213-2600(15)00545-7
39. Stallings VA, Sainath N, Oberle M, Bertolaso C, Schall JI. Energy balance and mechanisms of weight gain with ivacaftor treatment of cystic fibrosis gating mutations. J Pediatr. (2018) 201:229–37.e4. doi: 10.1016/j.jpeds.2018.05.018
40. Sun X, Yi Y, Yan Z, Rosen BH, Liang B, Winter MC, et al. In utero and postnatal VX-770 administration rescues multiorgan disease in a ferret model of cystic fibrosis. Sci Transl Med. (2019) 11:eaau7531. doi: 10.1126/scitranslmed.aau7531
41. Fortner CN, Seguin JM, Kay DM. Normal pancreatic function and false-negative CF newborn screen in a child born to a mother taking CFTR modulator therapy during pregnancy. J Cyst Fibros. (2021) 20:835–6. doi: 10.1016/j.jcf.2021.03.018
42. Gould MJ, Smith H, Rayment JH, Machida H, Gonska T, Galante GJ. CFTR modulators increase risk of acute pancreatitis in pancreatic insufficient patients with cystic fibrosis. J Cystic Fibrosis. (2021). doi: 10.1016/j.jcf.2021.09.010. [Epub ahead of print].
43. Megalaa R, Gopalareddy V, Champion E, Goralski JL. Time for a gut check: pancreatic sufficiency resulting from CFTR modulator use. Pediatr Pulmonol. (2019) 54:E16–8. doi: 10.1002/ppul.24353
44. Akshintala VS, Kamal A, Faghih M, Cutting GR, Cebotaru L, West NE, et al. Cystic fibrosis transmembrane conductance regulator modulators reduce the risk of recurrent acute pancreatitis among adult patients with pancreas sufficient cystic fibrosis. Pancreatology. (2019) 19:1023–6. doi: 10.1016/j.pan.2019.09.014
45. Ramsey ML, Gokun Y, Sobotka LA, Wellner MR, Porter K, Kirkby SE, et al. Cystic fibrosis transmembrane conductance regulator modulator use is associated with reduced pancreatitis hospitalizations in patients with cystic fibrosis. Am J Gastroenterol. (2021) 116:2446–54. doi: 10.14309/ajg.0000000000001527
Keywords: cystic fibrosis, exocrine pancreas, CFTR modulators, pancreatitis, precision medicine
Citation: McKay IR and Ooi CY (2022) The Exocrine Pancreas in Cystic Fibrosis in the Era of CFTR Modulation: A Mini Review. Front. Pediatr. 10:914790. doi: 10.3389/fped.2022.914790
Received: 07 April 2022; Accepted: 11 May 2022;
Published: 27 June 2022.
Edited by:
Emily Perito, University of California, San Francisco, United StatesReviewed by:
Kelvin D. MacDonald, Oregon Health and Science University, United StatesCopyright © 2022 McKay and Ooi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chee Y. Ooi, a2VpdGgub29pQHVuc3cuZWR1LmF1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.