 Yueerlanmu Tuoheti
Yueerlanmu Tuoheti- 1Medical School of Nanjing University, Nanjing, China
- 2Department of Gastroenterology, Children’s Hospital of Nanjing Medical University, Nanjing, China
Introduction: Transient Pseudohypoaldosteronism (TPHA) is a very rare condition usually secondary to urinary tract malformations (UTM) and/or urinary tract infection (UTI). It is characterized by hyperkalemia, hyponatremia, metabolic acidosis, and elevated plasma aldosterone levels. Given that the predominant manifestations of TPHA patients are digestive tract symptoms, such as poor appetite, vomiting, and weight gain, it is easily misdiagnosed as digestive tract diseases.
Case reports: Two children with poor appetite and vomiting were admitted to the Department of Gastroenterology, Children’s Hospital of Nanjing Medical University, from 2020 to 2021. Laboratory test results of these two children revealed hyponatremia (< 135.00 mmol/L), hyperkalemia (> 5.50 mmol/L), and hyperaldosteronism (> 180.00 ng/L). Moreover, genetic tests demonstrated no genetic variants highly associated with the phenotype in both cases. The two patients were subsequently treated with electrolyte correction. One of them also treated with antibiotics and one of them underwent surgery. They were followed for 8 and 4 months, respectively. No complications were observed during the follow-up period. This review aimed to outline both cases with parental consent.
Conclusion: Transient pseudohypoaldosteronism should be considered in children younger than 6 months, presenting with vomiting, poor appetite, unexplained hyponatremia, hyperkalemia, elevated aldosterone levels, and urethral malformation or urinary tract infection. Furthermore, attention should be paid to whether salt supplementation or anti-infection therapy is effective.
Introduction
Transient pseudohypoaldosteronism (TPHA) is characterized by hyperkalemia, hyponatremia, and metabolic acidosis associated with high levels of plasma aldosterone and renin, chiefly secondary to urinary tract malformation (UTM) and/or urinary tract infection (UTI) (1, 2). Since the first TPHA case was described in 1983, 149 TPHA-related cases have been reported abroad, whereas only 1 case has been documented in China (1). TPHA is easily misdiagnosed as a digestive tract disease, considering most cases are characterized by digestive tract symptoms, such as nausea, vomiting, sudden cardiac arrest, acute renal damage, and adrenal crisis (1, 3). Therefore, prompt diagnosis and early treatment are vital for the prognosis of children with TPHA.
Case reports
Case 1
A 2-month-old full-term female baby was admitted to our hospital with a one-month history of poor feeding and weight loss. She has experienced increased irritability, frequent emesis and lost 0.5 kg in the past month. Prior to her arrival at our hospital, she was prescribed oral probiotics (Bifidobacterium bifidum 0.5 g/kg/day for 7 days). Her birth and family history were unremarkable. On admission, her weight was 4 kg (−3 SD). Her mental and nutritional status was poor. Abdominal examination revealed a palpable mass of 3 cm × 4 cm in the upper left abdomen. She was administered 10% calcium gluconate (1 ml/kg/day IV for 4 day) and furosemide (1 mg/kg/day IV for 3 days) to lower serum potassium levels, 5% sodium bicarbonate (1.5 ml/kg/day IV for 3 days) to treat metabolic acidosis, and 0.9% sodium chloride (19743.75 mEq/kg/day IV) to address hyponatremia. After 8 days of treatment, her diet and electrolyte levels improved.
At the age of 3 and 5 months, she was hospitalized twice due to recurrent poor appetite. She was given hydrocortisone sodium succinate (32.62 mg/m2/day) and 9α-fludrocortisone (0.1 mg/day). The dose of hydrocortisone sodium succinate was gradually decreased, in addition to potassium reduction and salt supplementation. However, serum aldosterone levels kept fluctuating. At the age of 6 months, her general status gradually improved, and hydrocortisone sodium succinate and 9α-fludrocortisone were discontinued. The child was diagnosed with TPHA based on the onset, clinical manifestations, and lab and imaging test results. She was followed up for 8 months after discharge. At 13 months, her body weight was 8.6 kg (−1 SD∼M), and her developmental milestones were comparable to toddlers of her age. Finally, serum sodium, potassium, and aldosterone levels were normal.
Details of the laboratory test results are presented in Table 1. Other laboratory results were consistently normal, including angiotensin II levels, cortisol, plasma rennin activity, adrenocorticotropic hormone (ACTH), cortisol, sex hormone, androstenedione, 17-hydroxyprogesterone (17-OHP), urinalysis, urine culture, and chromosome analysis. The Genetic testing results collected from the medical record system showed a heterozygous variant (CPT2 gene c.1891 > T) was derived from the mother. However, we found no genetic variants highly associated with the phenotype. Electrocardiogram and pylorus ultrasonography findings were normal; abdominal CT scan revealed left retroperitoneal cystic foci and strong echo mass in the gallbladder in addition to abnormalities in the left kidney and ureter (Figure 1). Magnetic resonance urography (MRU) illustrated posterior renal cystic lesions associated with left calyces, left hydronephrosis, and left tortuous dilation of the ureter (with distal ureteral obstruction).
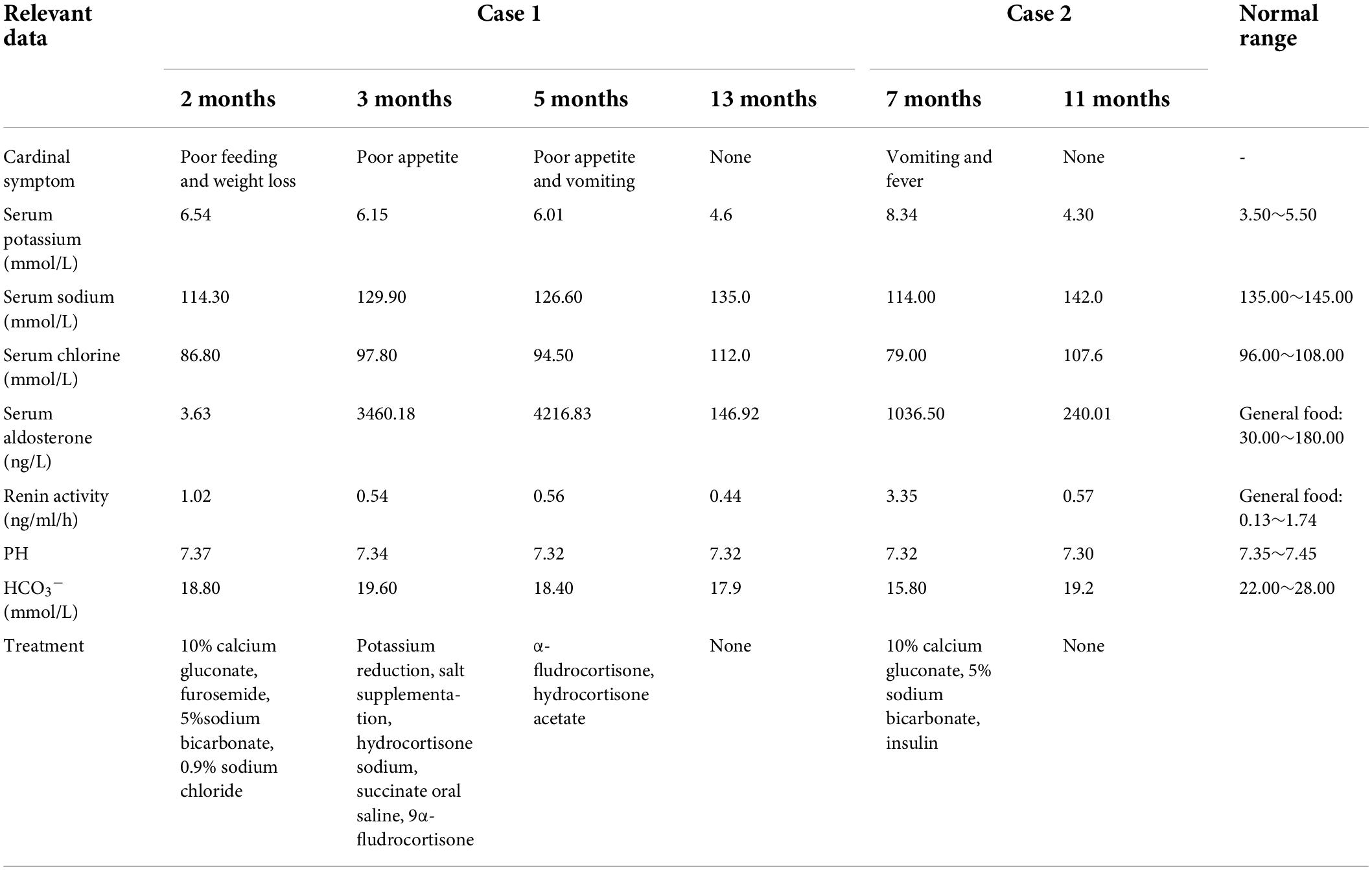
Table 1. Timeline with relevant data of the two transient pseudohypoaldosteronism cases.
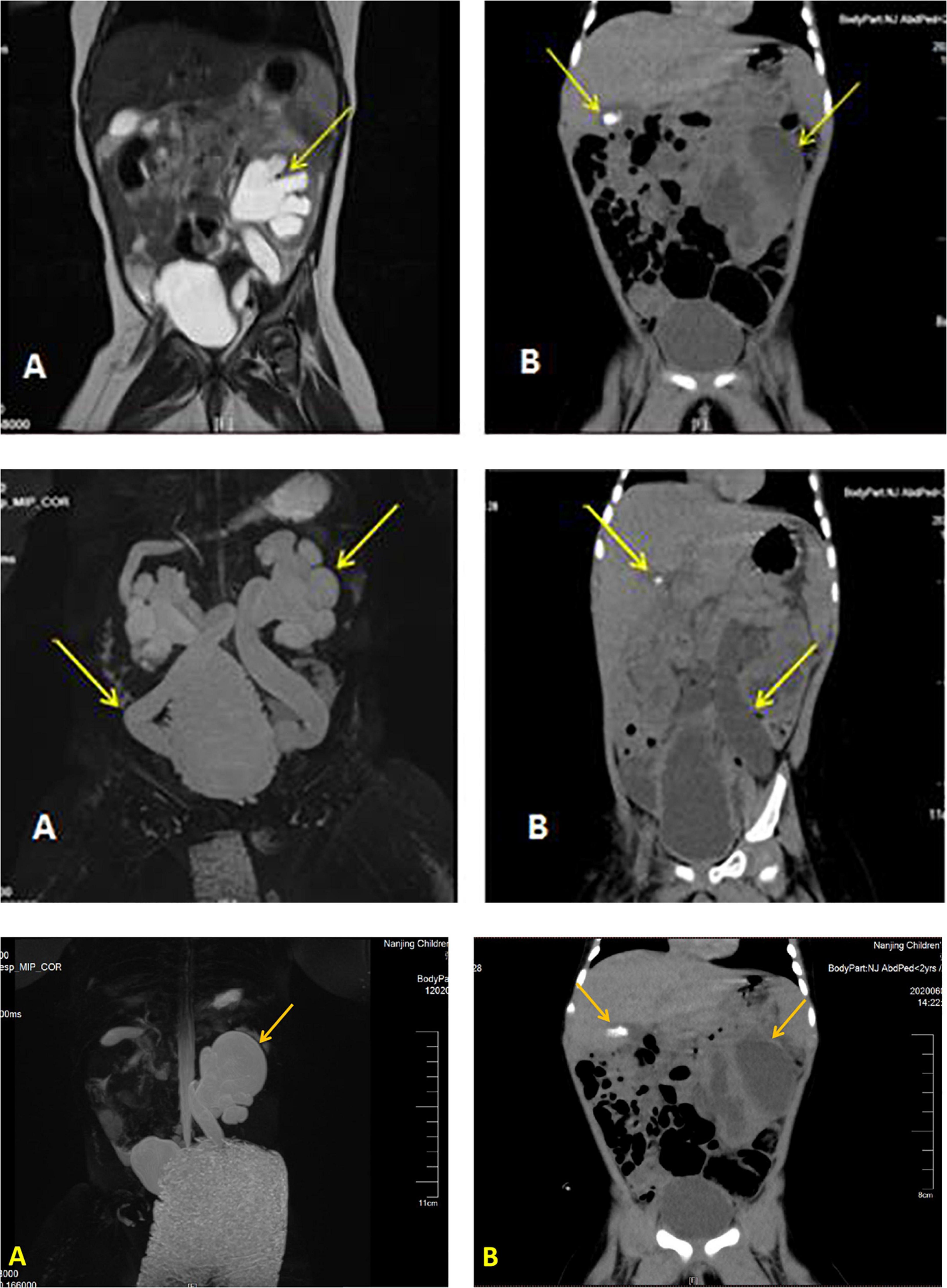
Figure 1. Imaging results displaying a dilated left ureter and renal pelvis and gallstone in the gallbladder. Image of the urinary MR of case 1 (A), Full abdominal CT of case 1 (B).
Case 2
A 7-month-old full-term male baby was admitted to our hospital with a 6–day history of vomiting and fever. The child received oral amoxicillin (150 mg/kg/day) in another hospital, but his condition did not improve. He had a history of bronchitis and recurrent eczema in the past 7 months, and his birth and family history were unremarkable. On admission, his body weight was 8 kg (−1SD∼M). His condition improved with the administration of 10% calcium gluconate (0.125 ml/kg/day IV single-dose), 5% sodium bicarbonate (0.75 ml/kg/day IV for 3 days), insulin (0.19 IU IH single-dose), sodium chloride supplementation (7678.12 mEq/kg/day PO and 9871.87 mEq/kg/day IV for 4 days), and intravenous meropenem (0.16 g/kg for 16 days). After 20 hours of treatment, serum potassium levels returned to normal, whereas hyponatremia persisted for 4 days. Following discharge, his plasma aldosterone level was marginally above average (240.01 ng/L). On the 19th day of admission, the patient underwent UTM correction (cystoscopy posterior urethral valvulotomy). To prevent reinfection, nitrofurantoin (1.56 mg/kg/day) was taken orally for 2 weeks, and the patient was followed up for 4 months. At 11 months, his weight was 11 kg, with normal motor development typical for a healthy child; indeed, he did not exhibit signs of TPHA during the 4-month follow-up.
His laboratory test results during hospitalizations are summarized in Table 1; the remaining aberrant laboratory results were as follows: angiotensin II 145.88 ng/L, cortisol 756.90 nmol/L, urine microalbumin 33.70 mg/L, urine leukocyte count 1321.30/μL, urine bacteria 2103.30/μL, Morganella morganella subspecies cultured in urine, CRP 98 mg/L, WBC 28.75 × 109/L, procalcitonin (PCT) 1.15 ng/mL, and HGB 118 g/L. Besides, expected laboratory results were sex hormones, ACTH levels, and kidney function indexes. The genetic test results of case 2 showed that he got heterozygote of these variants: c.1968 + 41G > A in the ABCB6 gene, c.1234C > G in the ARMC5 gene, c.4898T > A in the COL4A3 gene, c.1809G > C in the GPD2 gene, c.1543C > T in the HNF1B gene, c.2392C > T in the MLL5 gene, c.1279 G > A in the TFAP2A gene, c.8764G > A in the TNXB gene, and c.1978A > G in the IGSF1 gene. We found no intentional variants in the genes related to primary PHA of the child, including ABCB6 SCNN1A, SCNN1B, SCNN1G, NR3C2, WNK1, WNK4, and CUL3 gene. Abdominal CT scan results displayed bilateral hydronephrosis with obstruction of the ureter close to the bladder and gallstone (Figure 2); urinary tract (MRI and MRU) imaging showed bilateral hydronephrosis with a dilated ureter, as well as vesicoureteral reflux.
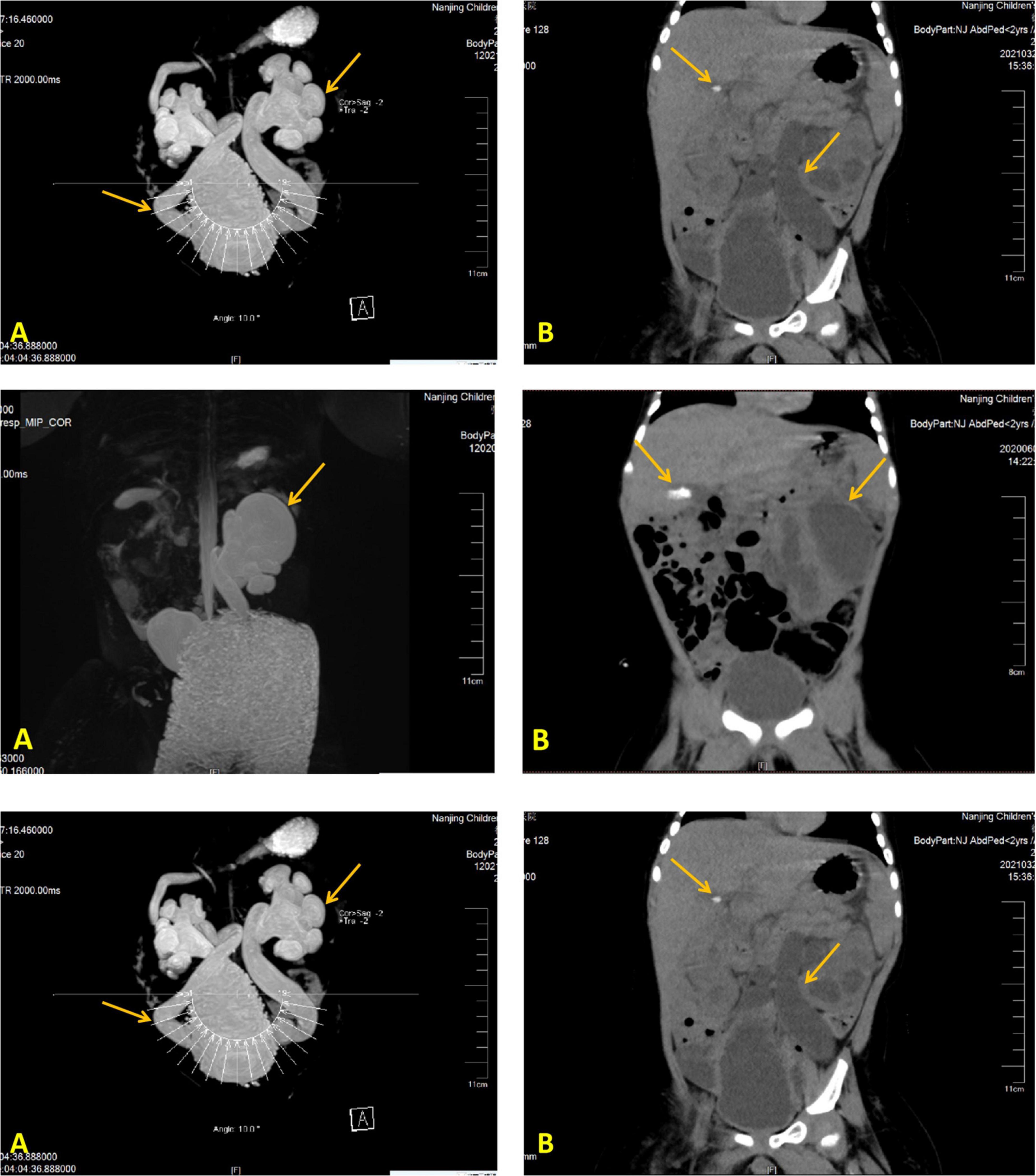
Figure 2. Imaging modalities and results illustrating a dilated left ureter and renal pelvis and gallstone in the gallbladder. Image of the urinary MR of case 2 (A), Full abdominal CT of case 2 (B).
Discussion
Pseudohypoaldosteronism (PHA) is a group of diseases characterized by electrolyte disorders, metabolic acidosis, and an elevated serum aldosterone level (1–3). It can be divided into primary pseudohypoaldosteronism and secondary pseudohypoaldosteronism (SPHA) according to etiology (2). The latter, also referred to as transient pseudohypoaldosteronism (TPHA), is a condition in which the renal tubule is unresponsiveness to aldosterone (1–3). Therefore, TPHA clinically manifests as transient and severe hyponatremia, hyperkalemia, metabolic acidosis, an elevated serum aldosterone level, and an elevated or normal renin activity (1–9).
It was previously established that TPHA in infants could be caused by multiple factors, including urinary tract malformations, urinary tract infection, and drugs (2, 10–14). Delforge et al. (3) described that TPHA caused by urethral malformation with urinary tract infection is more prevalent, but TPHA caused by unilateral UTM without UTI was rarely encountered. In case 2, bilateral ureteral obstruction was present, bacteria were cultured in the urine sample, and increased CRP levels and PCT and WBC counts implied the presence of a urinary tract infection. Therefore, case 2 was identified as TPHA resulting from bilateral UTM and UTI. Case 1 was considered a case of TPHA induced by unilateral urinary tract malformations (UTM) since results of imaging tests displayed distal obstruction of the left ureter with pyelogenic cysts, and there was no evidence of urinary tract infection.
The pathogenesis of TPHA remains ambiguous, but three factors, including age, urinary tract obstruction, and urinary tract infection, may be the keys to the development of TPHA (1, 3, 11). (1) Studies have validated that TPHA is more common in infants younger than 6 months, which may be related to their immature renal tubules (3, 15–17). (2) In children with UTM, urinary tract obstruction or vesicoureteral reflux leads to increased intrarenal pressure, which results in the down-regulation of aldosterone receptors (2, 5, 14). These two factors also lead to a surge in the intrarenal synthesis of various cytokines, such as tumor necrosis factor-β1 (TGF-β1), tumor necrosis factor-α (TNF-α), etc. The former can inhibit the action of aldosterone (3, 5, 15, 18, 19). (3) In children with UTI, internal and external toxins not only directly damage aldosterone receptors but also stimulate the immune system to generate inflammatory factors, such as interleukin-1 (IL-1), thromboxane, natriuretic peptide, etc. (15, 16, 18). TPHA has various clinical manifestations, most prominently manifesting as digestive symptoms, including poor feeding, vomiting, abdominal distension, and diarrhea (1, 13, 15). Children with TPHA may develop adrenal crisis, acute renal failure, and cardiac arrest associated with severe electrolyte disturbances (1, 12, 16, 18). Furthermore, researchers have reported cases of TPHA combined with pneumothorax and cholecystolithiasis. The pathogenesis of pneumothorax is still elusive, whereas cholecystolithiasis may be linked to chronic dehydration in children (1, 16). Herein, CT of the whole abdomen suggested that the two cases were complicated with gallstone.
Clinically, the symptoms of children with TPHA are comparable to sepsis or pyloric stenosis, but the differential diagnosis is straightforward, and the workup includes blood culture and pyloric ultrasound (5, 13). Nonetheless, several diseases, such as primary PHA, congenital adrenal hyperplasia (CAH), and congenital adrenal hypoplasia, typically present as severe and persistent hyponatremia, hyperkalemia, and metabolic acidosis and are clinically challenging to differentiate from TPHA (2, 8, 18, 20). Chromosome analysis and relevant genetic testing can be employed to identify this disease.
Transient pseudohypoaldosteronism treatment focuses on correcting electrolyte imbalance and acidosis, followed by the administration of antibiotics, correcting malformations, and addressing complications (3, 19). In children with a salt loss crisis, 9α-fluorohydrocortisone and hydrocortisone can be given prior to the diagnosis being made (2, 21). The administration of sodium, potassium, and UTM/TUI correction is effective in the majority of infants. Consequently, it is necessary to regularly monitor aldosterone levels, serum electrolytes, blood pH, and body weight and follow up for at least 1 year (2, 12, 19).
Conclusion
In summary, this study reported two cases of TPHA with vomiting and loss of appetite as the chief manifestations. TPHA should be considered in children under 6 months of age presenting with symptoms of vomiting, poor appetite, and weight loss. Finally, serum electrolyte levels, urinary tract ultrasound, and genetic testing should be performed on these patients.
Data availability statement
The original contributions presented in this study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Children’s Hospital of Nanjing Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
YT and YZ contributed to the study design, data collection, analysis, and manuscript preparation. YL contributed to data analysis, manuscript preparation, and revised the manuscript. ML and YJ contributed to the study design and critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Watanabe T, Yamazaki A. Pneumothorax and transient pseudohypoaldosteronism in an infant with hydronephrosis. Pediatr Nephrol. (2003) 18:62–4. doi: 10.1007/s00467-002-1005-0
2. Korkut S, Akin L, Hatipoglu N, Özdemir A, Gündüz Z, Dursun I, et al. A potential serious complication in infants with congenital obstructive uropathy: secondary pseudohypoaldosteronism. J Pak Med Assoc. (2019) 69:108–12.
3. Delforge X, Kongolo G, Cauliez A, Braun K, Haraux E, Buisson P. Transient pseudohypoaldosteronism: a potentially severe condition affecting infants with urinary tract malformation. J Pediatr Urol. (2019) 15:265.e1–7. doi: 10.1016/j.jpurol.2019.03.002
4. Nissen M, Dettmer P, Thränhardt R, Winter K, Niemeyer T, Tröbs RB. Congenital jejunal membrane causing transient pseudohypoaldosteronism and hypoprothrombinemia in a 7-week-old infant. Klin Padiatr. (2017) 229:302–3. doi: 10.1055/s-0043-113570
5. Abraham M-B, Larkins N, Choong C-S, Shetty V-B. Transient pseudohypoaldosteronism in infancy secondary to urinary tract infection. J Paediatr Child Health. (2017) 53:458–63. doi: 10.1111/jpc.13481
6. Krishnappa V, Ross JH, Kenagy DN, Raina R. Secondary or transient pseudohypoaldosteronism associated with urinary tract anomaly and urinary infection: a case report. Urol Case Rep. (2016) 8:61–2. doi: 10.1016/j.eucr.2016.07.001
8. Nandagopal R, Vaidyanathan P, Kaplowitz P. Transient pseudohypoaldosteronism due to urinary tract infection in infancy: a report of 4 cases. Int J Pediatr Endocrinol. (2009) 2009:195728. doi: 10.1155/2009/195728
9. Rodríguez-Soriano J, Vallo A, Oliveros R, Castillo G. Transient pseudohypoaldosteronism secondary to obstructive uropathy in infancy. J Pediatr. (1983) 103:375–80. doi: 10.1016/s0022-3476(83)80406-5
10. Kumar S, McDermott H, Kamupira S, Agwu JC. Rare case of pseudohypoaldosteronism in a neonate secondary to congenital hydrometrocolpos. BMJ Case Rep. (2020) 13:e234813. doi: 10.1136/bcr-2020-234813
11. Sethi S-K, Wazir S, Bansal S, Khokhar S, Wadhwani N, Raina R. Secondary pseudohypoaldosteronism masquerading congenital adrenal hyperplasia in a neonate. Kidney Int Rep. (2018) 3:752–4. doi: 10.1016/j.ekir.2018.01.004
12. Fujinaga S, Ohtomo Y, Someya T, Shimizu T, Yamashiro Y. Transient pseudohypoaldosteronism complicating acute renal failure in an infant with vesico-ureteral reflux and pyelonephritis. Pediatr Int. (2009) 51:744–6. doi: 10.1111/j.1442-200X.2009.02900.x
13. Maruyama K, Watanabe H, Onigata K. Reversible secondary pseudohypoaldosteronism due to pyelonephritis. Pediatr Nephrol. (2002) 17:1069–70. doi: 10.1007/s00467-002-0993-0
14. Pumberger W, Frigo E, Geissler W. Transient pseudohypoaldosteronism in obstructive renal disease. Eur J Pediatr Surg. (1998) 8:174–7. doi: 10.1055/s-2008-1071148
15. Kaninde A, Grace M-L, Joyce C, Taylor N-F, Ghataore L, Riordan M-F, et al. The incidence of transient infantile pseudohypoaldosteronism in Ireland: a prospective study. Acta Paediatr. (2021) 110:1257–63. doi: 10.1111/apa.15688
16. Kibe T, Sobajima T, Yoshimura A, Uno Y, Wada N, Ueta I. Secondary pseudohypoaldosteronism causing cardiopulmonary arrest and cholelithiasis. Pediatr Int. (2014) 56:270–2. doi: 10.1111/ped.12267
17. Martinerie L, Pussard E, Foix-L’Hélias L, Petit F, Cosson C, Boileau P, et al. Physiological partial aldosterone resistance in human newborns. Pediatr Res. (2009) 66:323–8. doi: 10.1203/PDR.0b013e3181b1bbec
18. Zhu Y, Kuang X-Y, Kang Y-L, Zhu G-H, Huang W-Y, Wu Y. Clinical analysis of an infant case with transient pseudohypoaldosteronism—salt-losing crisis. J Clin Pediatr. (2019) 37:601–4. doi: 10.3969/j.issn.1000-3606.2019.08.011
19. Bülchmann G, Schuster T, Heger A, Kuhnle U, Joppich I, Schmidt H. Transient pseudohypoaldosteronism secondary to posterior urethral valves–a case report and review of the literature. Eur J Pediatr Surg. (2001) 11:277–9. doi: 10.1055/s-2001-17151
20. De Clerck M, Vande Walle J, Dhont E, Dehoorne J, Keenswijk W. An infant presenting with failure to thrive and hyperkalaemia owing to transient pseudohypoaldosteronism: case report. Paediatr Int Child Health. (2018) 38:277–80. doi: 10.1080/20469047.2017.1329889
Keywords: digestive tract symptom, electrolyte disturbances, transient pseudohypoaldosteronism, urinary tract infection, urinary tract abnormalities
Citation: Tuoheti Y, Zheng Y, Lu Y, Li M and Jin Y (2022) Transient pseudohypoaldosteronism in infancy mainly manifested as poor appetite and vomiting: Two case reports and review of the literature. Front. Pediatr. 10:895647. doi: 10.3389/fped.2022.895647
Received: 14 March 2022; Accepted: 25 July 2022;
Published: 25 August 2022.
Edited by:
Arvind Bagga, All India Institute of Medical Sciences, IndiaReviewed by:
Vandana Jain, All India Institute of Medical Sciences, IndiaEmanuele Pignatti, University of Bern, Switzerland
Copyright © 2022 Tuoheti, Zheng, Lu, Li and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mei Li, limei6389@aliyun.com; Yu Jin, jinyuldyy@163.com
†These authors have contributed equally to this work