
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 17 August 2022
Sec. Pediatric Nephrology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.890147
 Yan Liu1†Wenchao Sheng2,3,4†Jinying Wu3,4†Jie Zheng3,4Xiufang Zhi2,3,4Shuyue Zhang2,3,4Chunyu Gu2,3,4Detong Guo2,3,4
Yan Liu1†Wenchao Sheng2,3,4†Jinying Wu3,4†Jie Zheng3,4Xiufang Zhi2,3,4Shuyue Zhang2,3,4Chunyu Gu2,3,4Detong Guo2,3,4 Wenhong Wang1*
Wenhong Wang1*Proximal renal tubular acidosis (pRTA) with ocular abnormalities is an autosomal recessive disease caused by variants in the Solute Carrier Family 4 Member 4 (SLC4A4) gene. Patients present with metabolic acidosis and low plasma bicarbonate concentration (3∼17 mmol/L). In addition, they are often accompanied by ocular abnormalities, intellectual disability, and growth retardation. The patient underwent whole exome sequencing (WES) and bioinformatics analysis of variant pathogenicity in this study. Then, a minigene assay was conducted to analyze the splicing site variant further. Compound heterozygous variants in the SLC4A4 gene (NM_003759.3), c.145C > T (p.Arg49*) and c.1499 + 1G > A, were detected by WES. The minigene assay showed an mRNA splicing aberration caused by the c.1499 + 1G > A variant. Compared with the wild type, the mutant type caused 4-base insertion between exons 10 and 11 of SLC4A4 after expression in HEK293 cells. In conclusion, the c.1499 + 1G > A variant in the SLC4A4 gene may be one of the genetic causes in the patient. Moreover, our study provides the foundation for future gene therapy of such pathogenic variants.
Proximal renal tubular acidosis (pRTA) with ocular abnormalities (OMIM 604278) is an autosomal recessive renal disease. The symptoms include renal tubular acidosis (RTA), ocular abnormalities (glaucoma, band keratopathy, and cataract), intellectual disability, and growth retardation. The disease is caused by variants in the Solute Carrier Family 4 Member 4 (SLC4A4) gene. The SLC4A4 gene spans approximately 450 kb and contains 26 exons (1); it is mapped to chromosome 4q13.3. The human SLC4A4 gene encodes electrogenic sodium bicarbonate cotransporter 1 (NBC1), mainly expressed in kidney, pancreas, brain, corneal, and other tissues (2). The protein’s primary function is to regulate the secretion and absorption of bicarbonate and regulate intracellular pH (3) (Figure 1A). According to the HGMD variant database, reported studies have found 17 nonsense/missense variants (Figure 1B). Furthermore, the clinical phenotype of patients with nonsense/missense variants is pRTA with ocular abnormalities (4).
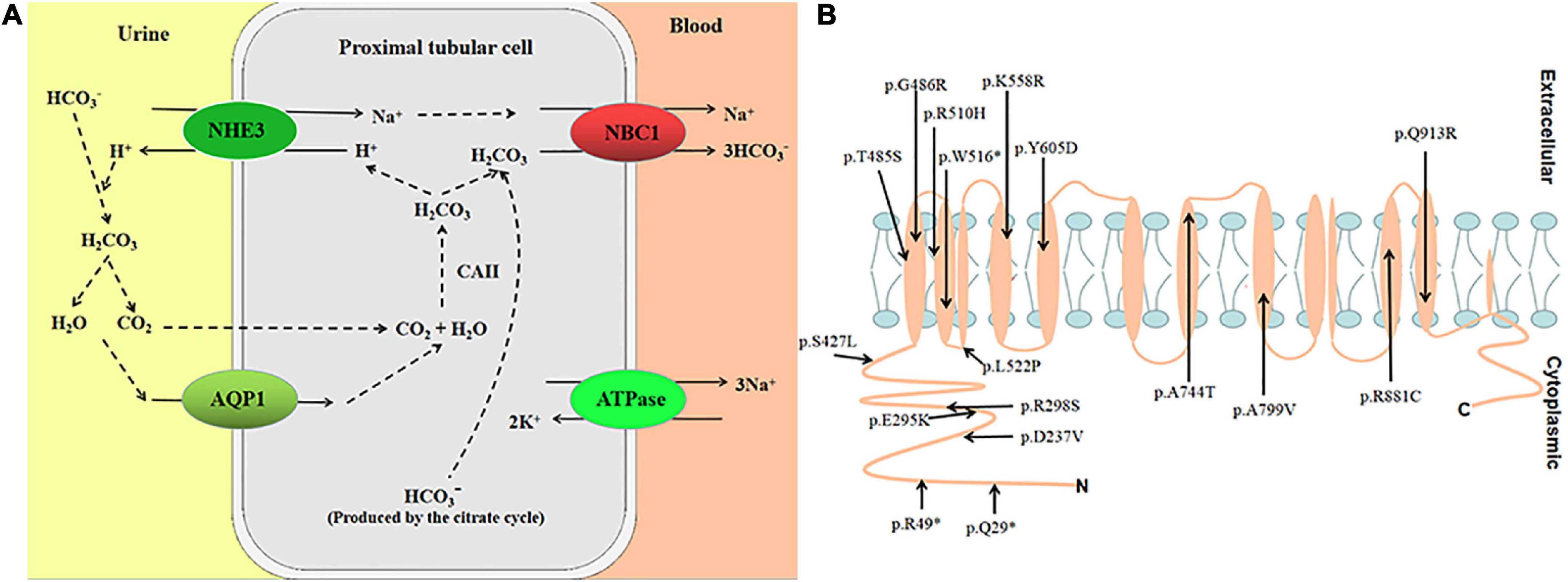
Figure 1. (A) Transepithelial HCO3– transport is mediated by the coupling of apical Na+/H+ exchange (NHE3) and basolateral NBC1 functioning as an electrogenic Na+-CO32–cotransporter with a 1:2 charge transport stoichiometry. (B) Putative location of variants in NBC1.
In this study, we found a Chinese patient with pRTA and compound heterozygous variants, c.145C > T (p.Arg49*) and c.1499 + 1G > A, in the SLC4A4 gene (NM_003759.3). Furthermore, in vitro functional studies were performed for the splice site variant c.1499 + 1G > A.
The whole exome sequencing (WES) of the patient was performed by the KingMed company (Guangzhou, China). Paired-end sequencing was performed in > 95% of target regions, including all coding regions, canonical splice sites, and exon-intron boundaries with reading lengths of 150 bp. Moreover, the sequencing had an average coverage depth of 100-fold. The Burrows-Wheeler Aligner software was used to map raw data reads to the human reference genome GRCh37/hg19. The Genome Analysis Toolkit software was also used to analyze insertion, deletion, and SNP sites. Variant annotations of the KMTD, HGMD, ClinVar, ESP6500, dbSNP, and 1,000 Genomes databases were added with the Annovar software. The classification of variants followed the American College of Medical Genetics and Genomics criteria (ACMG) (5).
Genomic DNA was extracted from the peripheral blood with Blood Genomic DNA Mini Kit (CWBIO, Beijing, China) according to the manufacturer’s instructions. Three fragments of the human SLC4A4 gene containing exons 9/10/11 of SLC4A4 and flanking intronic regions were amplified by PCR using a Taq Master Mix dye (CWBIO, Beijing, China) and primers were engineered to have restriction sites (Supplementary material). The PCR products were separated on 1% agarose gel. The gel was purified, purified DNAs were used as templates, and the recombinant plasmid was constructed with pcDNA3.1 and the templates. Then, the constructed recombinant plasmid was sequenced, and verified clones were referred to as the wild type (WT). We obtained mutant type (MUT) clones by Fast Site-Directed Mutagenesis Kit (TIANGEN, Beijing, China). The primers (F-primer: 5′-TCTATTTAATTTCAGCAAATATGTACATACAT-3′, R-pri mer: 5′-TTTGCTGAAATTAAATAGA AGCCTCTCAAAA AC-3′) were used.
HEK293T cells from ZQXZBIO (Shanghai, China) were cultured in high-glucose Dulbecco’s modified eagle medium (Gibco, United States) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 mg/ml streptomycin at 37 and a humidified 5% CO2 incubator. The cells were cultured 24 h before transient transfection in six-well plates. Whereafter, the cells were transfected with the pcDNA3.1-SLC4A4 minigene (WT, MUT) using the Lipofectamine® 2000 transfection reagent (Thermo Fisher Scientific, United States). Thirty hours after transfection, the cells were harvested and suspended in TRIZOL (TIANGEN, Beijing, China) for total RNA extraction. Then, RNA was reverse-transcribed to cDNA using the FastKing RT Kit (with gDNase) (TIANGEN, Beijing, China) according to the manufacturer’s instructions. The cDNA was amplified using the primer set (F-primer: 5′-GTTCTGTGGTGGACTAATTA-3′, R-primer: 5′-CTGGGTCAGGTGGCACACA-3′). Lastly, RT-PCR products were separated and analyzed on a 1% agarose gel, visualized with DNA ladder and ethidium bromide, gel-purified, and sequenced.
To predict the effect of the variant on pre-mRNA splicing, an in silico analysis was performed using Spliceman (6), SpliceAid2 (7), NetGene2 (8), BDGP (9), SROOGLE (10), HEX (11), RBP_map (12), MaxEnt (13), CRYR-SKIP (14), and SpliceAI (15).
The clinical symptoms of the patient have been previously reported (4). In brief, the patient presented with hypokalemia and general weakness 3 months before hospitalization. Besides, severe acidosis, short stature, epilepsy, encephalopathy, and disturbance of consciousness were shown. Both of the vitreous bodies were cloudy. The head MRI showed high-signal intensity in the bilateral cerebral cortex and symmetrical calcification spots in the bilateral basal ganglia. Therefore, the patient was suspected of having primary pRTA.
The compound heterozygous variants c.145C > T (p.Arg49*) and c.1499 + 1G > A were found in the SLC4A4 gene (NM_003759.3), but no other variants in the known pRTA pathogenic gene were detected. The variant c.145C > T was inherited from the healthy father, and the variant c.1499 + 1G > A was inherited from the healthy mother. The c.1499 + 1G > A variant had not been included in the HGMD database, which had zero frequency in the 1,000 Genomes Project database.
A minigene analysis was conducted to analyze the splicing site variant further. The SLC4A4 minigene structure was designed to study the effect of the c.1499 + 1G > A splice-site variant on pre-mRNA splicing (Figure 2A). The results of the RT-PCR products on agarose gel electrophoresis showed a single band in the WT and one band in the MUT. Moreover, the electropherogram showed the DNA fragments represented by the two strips might be similar in length (Figure 2B). The Sanger sequencing showed that WT cDNA-amplified products were consistent with SLC4A4 exons 9–11. While comparing with SLC4A4 exons 9–11, we found a 4-base insertion between exons 10 and 11 in MUT cDNA-amplified products (Figure 2C). The minigene analysis showed that the c.1499 + 1G > A variant could abrogate the intron 10 donor splice site. At the same time, the downstream cryptic donor splice site was activated, resulting in the 4 bases (ATAT) of intron 10 getting inserted between exons 10 and 11.
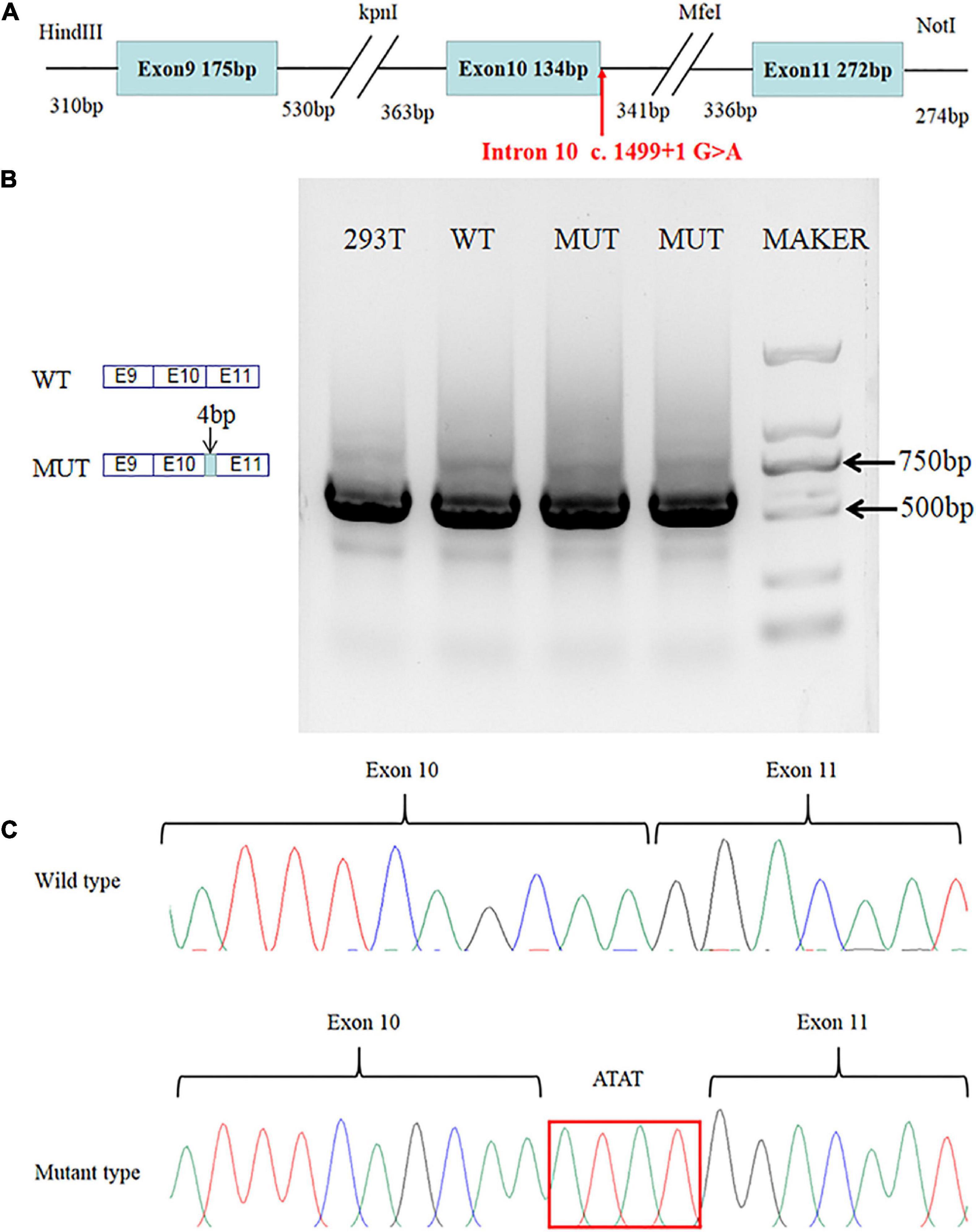
Figure 2. Results of the minigene assay. (A) SLC4A4 minigene structure: schematic diagram of the SLC4A4 minigene construct containing exon 9, part of intron 9, exon 10, part of intron 10, exon 11, and part of intron 11. (B) Agarose gel electrophoresis showed a single band in the WT and one band from the MUT. Moreover, the DNA fragments represented by the two strips might be similar in length. (C) The mutant type causes a 4-base insertion between exons 10 and 11 of SLC4A4 after expression in HEK293 cells.
Spliceman predicted that the splice site variant would lead to aberrant splicing with an L1 distance of 36,609 and a prediction score of 76%. Variant sequences had significantly lower splice donor scores than wild-type sequences: SROOGLE, 0 vs. 78.15; BDGP, 0 vs. 94; HEX, –4.95 vs. 1.02; MaxEnt, –0.74 vs. 7.44. The SpliceAid2 prediction showed the abnormal position of protein binding to the 5′ splice site (Supplementary material). Moreover, SpliceAI also showed loss of the 5′ splice site. No difference between the WT and the MUT were found with NetGene2 and CRYR-SKIP (Table 1). In addition, the variant transcript sequence was compared with the wild-type transcript sequence with GeneRunner (v6.5.52); the predicted results were splice site variants causing a frameshift.
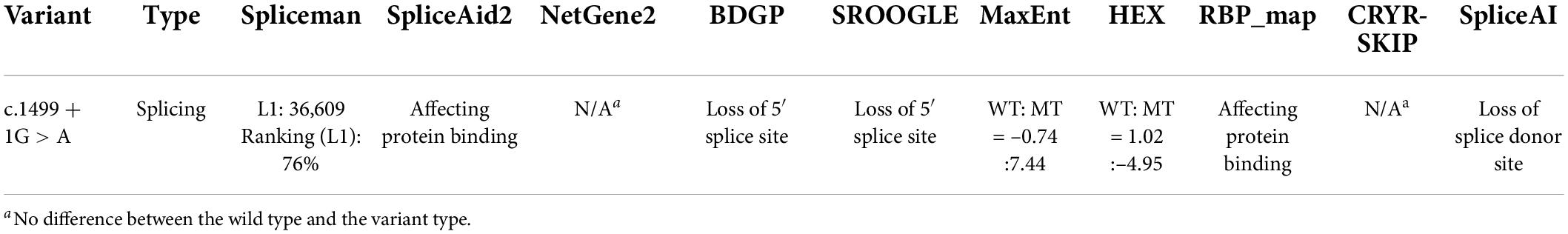
Table 1. In silico analysis prediction results of the variants in the SLC4A4 gene.
In this study, the compound heterozygous variants of the SLC4A4 gene were found by WES. Then, we further explored the splicing effect of the c.1499 + 1G > A variant. The pre-mRNA splicing mechanism suggests that the spliceosome performs removal of introns. In the process of intron recognition, U1 snRNP recognizes and binds to the AG-GU sequence at the donor splice site. In addition, U2snRNA is combined with a branch point sequence (16). Single nucleotide substitutions at the 5′ or 3′ canonical splice site are the most common splicing variants, leading to activated cryptic splice site, skip exon, or less intron retention (17). The c.1499 + 1G > A variant changes the 5′splice site sequence from 5′-CAAGTAT-3′ to 5′-CAAATAT-3′. It prevents proper recognition of intron 10 by the spliceosome, resulting in a few intron sequence retentions immediately downstream of the variant donor splice site during pre-mRNA splicing. The sequencing results show that the sequence (ATAT) is retained between exons 10 and 11, which caused a frameshift. It is worth noting in the study that the RNA expressed by the SLC4A4 gene was not detected in the patient′s or his parents’ peripheral blood samples, and kidney tissue cells were unavailable. Therefore, we tried to evaluate the impact of splicing by minigene assay in vitro. This method provides a new idea for studying the mechanism of splicing abnormalities when patient samples are not available or genes do not express in peripheral blood (18).
However, in this study, the splicing site information that BDGP predicted was not consistent with in vitro experiments (Table 2). SROOGLE showed loss of the 5′ splice site only but did not provide new splice site information. By splicing site analysis, programs can predict whether the splice site is altered, but they are not wholly accurate. In this study, we learn that minigene assay can clarify the mechanism of splicing abnormalities, provide a higher level of pathogenic evidence (PS3) for variants, and improve the pathogenicity classification level of variants. In addition, we further speculated how the cryptic splice site was activated. The prediction of RNA secondary structure by mFold (19) found no difference between the optimal predicted structures of the wild type and the mutant type, but the 5′ donor splice site of intron 10 and cryptic splice sites were exposed in the same hairpin loop region (Supplementary material). Combining the results of the above minigene assay and SpliceAid2 predictions, we consider that one of the reasons for the activation of the cryptic splice site may be that the c.1499 + 1G > A variant makes the normal donor splice site disappear. The cryptic splice site in the hairpin loop becomes particularly exposed, and the spliceosome recognizes cryptic splice sites leading to splicing abnormalities.

Table 2. Results of BDGP and minigene assay.
In conclusion, the c.1499 + 1G > A in the SLC4A4 gene is a null variant (PVS1). In vitro or in vivo functional studies on the c.1499 + 1G > A variant demonstrates that the variant has a damaging effect (PS3). c.1499 + 1G > A is absent from controls in 1,000 Genomes Project (PM2). For recessive disorders, they are detected in trans with a pathogenic variant (PM3). Multiple lines of computational evidence support a deleterious effect on the gene (PP3). According to the ACMG guidelines, the variant is classified as “pathogenic” based on the above evidence. In addition, the clinical manifestation of pRTA with ocular abnormalities is severe, and the prognosis is poor. Prenatal diagnosis and genetic counseling are vital for such disease. Early identification of pathogenic variant is the basis of prenatal diagnosis and genetic counseling. Our study is of great significance to prenatal diagnosis and prevention of birth of children with the disease. Moreover, the therapeutic potential of antisense oligonucleotides (ASOs) for correcting splicing defects that cause genetic diseases has been investigated. It was reported that ASOs could improve the abnormal splicing effect of the c.6937 + 594T > G variant in the BRCA2 gene to a certain extent (20). Another study showed that ASOs blocked the aberrant splicing created by CFTR c.3718-2477C > T mutation, redirected the splicing to the correct splicing site, and restored chloride secretion in bronchial epithelial cells of patients with cystic fibrosis (21). Our study provides the basis for future gene therapy of such pathogenic variants.
In conclusion, SLC4A4 variants, including c.1499 + 1G > A and c.145C > T (p.Arg49*), may be the genetic basis of the patient. Moreover, the splicing abnormality is verified in vitro, and this discovery will contribute to accurate diagnosis. Further functional studies may help reveal the potential mechanism of SLC4A4 gene variants causing pRTA with ocular abnormalities and provide possible gene therapy in the future.
The original contributions presented in this study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the Ethical Committee of Tianjin Children’s Hospital. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
YL, WS, and WW designed the experiments. YL, WS, JW, JZ, and SZ performed the experiments. WS wrote the manuscript. WW, SZ, XZ, DG, JZ, and CG provided revisions and edits of the manuscript. All authors commented on previous versions of the manuscript and read and approved the final version of the manuscript.
This study was supported by the Public Health and Technology Project of Tianjin and Key Projects of Tianjin Children’s Hospital (grant nos. TJWJ2021ZD007 and Y2020003).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.890147/full#supplementary-material
1. Abuladze N, Song M, Pushkin A, Newman D, Lee I, Nicholas S, et al. Structural organization of the human NBC1 gene: kNBC1 is transcribed from an alternative promoter in intron 3. Gene. (2000) 251:109–22. doi: 10.1016/s0378-1119(00)00204-3
2. Burnham CE, Amlal H, Wang Z, Shull GE, Soleimani M. Cloning and functional expression of a human kidney Na+:HCO3- cotransporter. J Biol Chem. (1997) 272:19111–4. doi: 10.1074/jbc.272.31.19111
3. Bernardo AA, Bernardo CM, Espiritu DJ, Arruda JA. The sodium bicarbonate cotransporter: Structure, function, and regulation. Semin Nephrol. (2006) 26:352–60. doi: 10.1016/j.semnephrol.2006.07.008
4. Liu Y, Wang WW, Lei MF, Chen WY. (Severe proximal renal tubular acidosis with ocular abnormalities caused by SLC4A4 gene variant: A case report). Zhonghua Er Ke Za Zhi. (2020) 58:241–2. doi: 10.3760/cma.j.issn.0578-1310.2020.03.018
5. Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the association for molecular pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. (2017) 19:4–23. doi: 10.1016/j.jmoldx.2016.10.002
6. Spliceman. Spliceman home page. (2021). Available online at: http://fairbrother.biomed.brown.edu/spliceman2/upload (accessed November 22, 2021).
7. SpliceAid 2. SpliceAid 2: A database of human splicing factors expression data and RNA target motifs. (2021). Available online at: http://193.206.120.249/splicing_tissue.html (accessed November 22, 2021).
8. NetGene2. NetGene2 intron splice sites in human, C. elegans and A. thaliana DNA. (2021). Available online at: http://www.cbs.dtu.dk/services/NetGene2/ (accessed November 22, 2021).
9. Berkeley Drosophila Genome Project [BDGP]. Berkeley drosophila genome project. (2021). Available online at: https://www.fruitfly.org/seq_tools/splice.html (accessed November 22, 2021).
10. Sroogle. Splicing regulation online graphical engine. (2021). Available online at: http://sroogle.tau.ac.il/ (accessed November 22, 2021).
11. HEXplorer Score. HEXplorer Score. (2021). Available online at: https://www2.hhu.de/rna/html/hexplorer_score.php (accessed November 22, 2021).
12. RBPmap. RBPmap Mapping Binding Sites of RNA Binding Proteins. (2021). Available online at: http://rbpmap.technion.ac.il/ (accessed November 22, 2021).
13. MaxEntScan. MaxEntScan::build for Short Sequence Motifs. (2021). Available online at: http://hollywood.mit.edu/burgelab/maxent/Xmaxent.html (accessed November 22, 2021).
14. CRYR-SKIP. CRYR-SKIP. (2021). Available online at: https://cryp-skip.img.cas.cz/ (accessed November 22, 2021).
15. Ensembl. Variant Effect Predictor-SpliceAI. (2021). Available online at: https://asia.ensembl.org/Homo_sapiens/Tools/VEP (accessed November 22, 2021).
16. Fredericks AM, Cygan KJ, Brown BA, Fairbrother WGRNA-. Binding proteins: Splicing factors and disease. Biomolecules. (2015) 5:893–909. doi: 10.3390/biom5020893
17. Pucker B, Brockington SF. Genome-wide analyses supported by RNA-Seq reveal non-canonical splice sites in plant genomes. BMC Genomics. (2018) 19:980. doi: 10.1186/s12864-018-5360-z
18. Nakajima Y, Meijer J, Zhang C, Wang X, Kondo T, Ito T, et al. Altered Pre-mRNA splicing caused by a novel intronic mutation c.1443+5G>A in the dihydropyrimidinase (DPYS) gene. Int J Mol Sci. (2016) 17:86. doi: 10.3390/ijms17010086
19. mFlod. (2021). Available online at: http://www.mfold.org/mfold/applications/rna-folding-form-v2.php. (accessed July 16, 2021).
20. Anczukow O, Buisson M, Leone M, Coutanson C, Lasset C, Calender A, et al. BRCA2 deep intronic mutation causing activation of a cryptic exon: Opening toward a new preventive therapeutic strategy. Clin Cancer Res. (2012) 18:4903–9. doi: 10.1158/1078-0432.CCR-12-1100
Keywords: compound heterozygous variants, SLC4A4, pre-mRNA, minigene assay, splicing
Citation: Liu Y, Sheng W, Wu J, Zheng J, Zhi X, Zhang S, Gu C, Guo D and Wang W (2022) Case report: Altered pre-mRNA splicing caused by intronic variant c.1499 + 1G > A in the SLC4A4 gene. Front. Pediatr. 10:890147. doi: 10.3389/fped.2022.890147
Received: 05 March 2022; Accepted: 25 July 2022;
Published: 17 August 2022.
Edited by:
Agnieszka Swiatecka-Urban, University of Virginia, United StatesReviewed by:
Emanuele Buratti, International Centre for Genetic Engineering and Biotechnology, ItalyCopyright © 2022 Liu, Sheng, Wu, Zheng, Zhi, Zhang, Gu, Guo and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenhong Wang, ZG9jd3doQDEyNi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.