
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 24 May 2022
Sec. Genetics of Common and Rare Diseases
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.887214
This article is part of the Research Topic Highly Consanguineous Populations and Rare Genetic Diseases in Pediatrics View all 11 articles
 Di Zhang1
Di Zhang1 Yi Qu1
Yi Qu1 Xue-Qi Dong1
Xue-Qi Dong1 Yi-Ting Lu1Kun-Qi Yang1
Yi-Ting Lu1Kun-Qi Yang1 Xin-Chang Liu1
Xin-Chang Liu1 Peng Fan1
Peng Fan1 Yu-Xiao Hu2Chun-Xue Yang2
Yu-Xiao Hu2Chun-Xue Yang2 Ling-Gen Gao3Ya-Xin Liu2*
Ling-Gen Gao3Ya-Xin Liu2* Xian-Liang Zhou1*
Xian-Liang Zhou1*Objective: Liddle syndrome (LS) is a monogenic hypertension consistent with autosomal dominant inheritance, often with early onset high blood pressure in childhood or adolescence. This study aimed to identify the pathogenicity of a nonsense mutation in SCNN1G in a Chinese family with LS and the long-term outcomes of tailored treatment with amiloride.
Methods: To explore the pathogenicity of candidate variant reported in 2015 by our team, we constructed mutant and wild-type models in vitro and measured amiloride-sensitive current in Chinese Hamster Ovary (CHO) cells using patch clamp technique. Participants were followed up for 7 years after tailored treatment with amiloride.
Results: A nonsense variant was detected in six members, two of whom were pediatric patients. This mutation resulted in a termination codon at codon 572, truncating the Pro-Pro-Pro-X-Tyr motif. The mutant epithelial sodium channels displayed higher amiloride-sensitive currents than the wild-type channels (P < 0.05). Tailored treatment with amiloride achieved ideal blood pressure control in all patients with normal cardiorenal function, and no adverse events occurred during follow-up.
Conclusion: We found the pathogenicity of a nonsense SCNN1G mutation (p.Glu571*) with enhanced amiloride-sensitive currents in a LS family with young patients. Tailored treatment with amiloride may be an effective strategy for the long-term control of blood pressure and protection from target organ damage or cardiovascular events, including children and youth patients with LS.
Liddle syndrome (LS) was first reported at 1963 by Liddle et al. (1). It is a monogenic hypertension caused by mutations in genes encoding epithelial sodium channels (ENaCs) (2). ENaCs are composed of three subunits: α, β, and γ (encoded by SCNN1A, SCNN1B, and SCNN1G, respectively) (3). Proline-rich segments, referred to as Pro-Pro-Pro-X Tyr (PY) motifs, in the carboxyl terminal regions of β and γ subunits are binding sites for WW domains of neural precursor cell expressed, developmentally Down-regulated 4 (NEDD4), an E3 ubiquitin-protein ligase (4). By binding the ubiquitin-protein ligase domain, PY motifs inactivate ENaCs (5). LS is characterized with increased sodium reabsorption in collecting tubules resulted from ENaCs dysfunction, including inactivation disorders, and abnormal opening frequencies (3, 6–8). The function of ENaCs usually is examined by patch clamp techniques.
The typical manifestations of LS include early-onset hypertension, hypokalemia and suppression of plasma renin activity (PRA), and plasma aldosterone concentration (PAC) (3, 9). Some LS patients lack a typical presentation and are not diagnosed and treated in time, which can lead to major complications such as stroke and early death because of poor control of hypertension (10–13). Genetic sequencing is recommended for LS screening for SCNN1A, SCNN1B, and SCNN1G mutations, especially in families with a history of early-onset hypertension (3). Through family screening, it often screens for children who carried mutations in family (11, 14). Genetic sequencing screened LS patients, but the pathogenicity of most variants has not been verified by functional experiments. For LS patients diagnosed based on sequencing results and silico analysis prediction, the usual follow-up after beginning amiloride treatment is approximately 1 or 2 months and the long-term outcomes of LS patient remains unclear.
In this study, we explored the pathogenicity of a nonsense mutation (c.1171G > T) in exon 13 of SCNN1G reported in 2015 year by our team and efficiency of blood pressure control by amiloride. We used patch-clamp to verify functional mechanism of this mutation, and found that amiloride-sensitive currents differed significantly between wild-type and mutant groups. All members harboring the c.1171 G > T mutation in SCNN1G with hypertension and/or abnormal biochemical presentations were diagnosed with LS. Over a 7-year follow-up, the results show efficacy with tailored treatment of amiloride in blood pressure control and prevention of target organ damage.
The index case was an 18-year-old woman (III-2) with early-onset, refractory, and unexplained hypertension and hypokalemia, accompanied by suppressed PRA. The diagnosis of hypertension for children is made when repeat BP values on three different measurements are greater than the 95th percentile for the age, sex, and height of the patient (15). The proband and five members carrying the nonsense mutation were included for the clinical, biochemical analysis in this family. This study was approved by the Ethics Committee of Fuwai Hospital and all participants signed their written informed consent (The informed consent of the deceased patient was obtained from his immediate family) in accordance with the Declaration of Helsinki.
All biochemical tests, such as of electrolytes, were performed using standard methods. Patients carrying SCNN1G mutation were required to follow a salt-restricted diet and received oral amiloride (5.0 mg/day for adults and 2.5 mg/day for children) intervention. Blood pressure and potassium levels were measured during follow-up period. Indicators of assessing cardiorenal function and cardiovascular events were followed up over 7 years. Indicators included creatinine, urea nitrogen, echocardiography, and cardiovascular events. Events contain heart failure, stroke, and all-cause death.
The sequence of human SCNN1A, SCNN1B, and SCNN1G was obtained from the University of California Santa Cruz database, and cDNAs encoding human ENaC were synthesized exogenously. The SCNN1G gene mutation (c.1171G > T) was induced using a QuikChange Site-Directed Mutagenesis Kit (Statagene). ENaC expression vectors were gifts from Dr. Cecilia Canessa, Yale University. Chinese Hamster Ovary (CHO) cells were used for expression human ENaC. CHO cells were cultured under standard conditions (10% fetal bovine serum in Dulbecco’s modified Eagle’s medium, 1% penicillin/streptomycin, 37°C, 5% CO2). Before transfection, cells were seeded on poly-lysine coated slides. pcDNA3.1-ENaCα, ENaCβ, ENaCγ, or ENaCγ-mut together with pEGFP-N1 were transfected into cells in a 1:1:1:1 ratio using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, United States). Green fluorescent protein was used to identify transfected cells. Amiloride (10 μM) was added to culture medium until just prior to the electrophysiological experiments (48 h after transfection).
Whole-cell current recordings were acquired using a MultiClamp 700B amplifier (Molecular Devices, San Jose, CA, United States). Signals were collected by a Micro 1401 MKII (Cambridge Electronic Design, Cambridge, United Kingdom) using Spike 2 acquisition software. The electrophysiological methods were as previously described (16). In brief, the pipette solution (in mM) was 120 CsCl, 5 NaCl, 5 EGTA, 2 MgCl2, 2 Mg-ATP, 0.1 GTP, and 10 HEPES (pH 7.4). The bath solution (in mM) was 150 NaCl, 1 CaCl2, 2 MgCl2, and 10 HEPES (pH 7.4). Whole-cell capacitance was recorded for normalizing ENaC currents. Current through ENaC was elicited by voltage ramping from 20 to -140 mV over a 300 ms period. At the end of each recording, 10 μM amiloride was added to identify amiloride-sensitive currents. ENaC activity was assessed as the amiloride-sensitive current.
We systematically searched MEDLINE (via PubMed), Embase, Cochrane, and Web of Science databases for relevant articles published in English up to December 31, 2020, with the search terms “Liddle syndrome,” “Liddle’s syndrome,” “pseudoaldosteronism,” and SCNN1G. Studies that reported patient clinical characteristics, outcomes as well as phenotype and genotype relation of Liddle syndrome patients carrying SCNN1G mutations and pathogenicity of variants were included.
Differences in amiloride-sensitive sodium currents between wild-type and mutant ENaCs in CHO cells were analyzed using the unpaired t-test. P-values less than 0.05 were considered statistically significant.
All members participating in this study presented with varying degree of hypertension. The echocardiographic findings of the proband (III-2) suggest concentric left ventricular hypertrophy, so it is presumed that hypertension occurred earlier than 18 years old. The I-1 and II-2 were paralyzed and bedridden from hypertensive cerebral hemorrhage prior the start of the study and the II-3 was diagnosed with hypertension and died of hypertensive stroke at 32 years of age. The IV-1, a 7 years old boy, was diagnosed with hypertension for BP values (120/72 mmHg) greater than 95th percentile reference (110/71 mmHg). All participants underwent biochemical examinations, and the results are shown in Table 1.
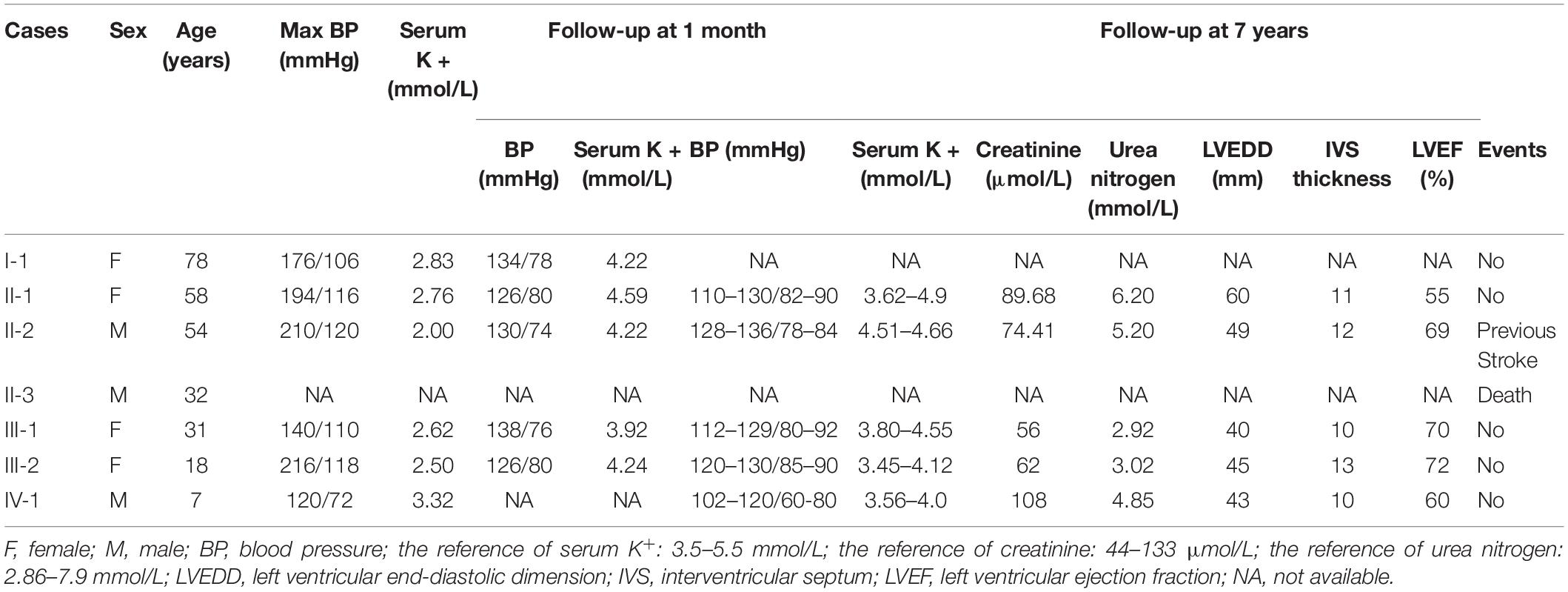
Table 1. Clinical, biochemical characteristics and results of follow-up of patients in this family.
The proband and other members diagnosed with LS were recommended to follow a low-salt diet and receive oral amiloride treatment. The blood pressures and serum potassium levels of all family members were in the normal range (Table 1) during follow-up period. Creatinine, urea nitrogen and the result of echocardiography were normal, and none of the family members had cardiovascular events during the 7-year period of regular treatment (Table 1).
A truncation mutation in exon 13 of SCNN1G was identified and reported in this family and this mutation led to a stop codon after glutamic acid (Glu) (p.Glu571*; c.1171G > T; Figure 1). We compared amiloride-sensitive sodium currents in p.Glu571* mutant and wild-type CHO cells. The amiloride-sensitive currents of the mutant control group were 3.7 times of the wild-type control group (3.7 vs. 1.0, P < 0.05; Figure 2A). After cells were incubated for 48 h after the addition of amiloride, we detected and compared the current changes in wild-type and mutant ENaCs cells. There was a significant difference between the mutant control and mutant amiloride-added groups (152.4 ± 41.6 pA/pF vs. 84.2 ± 26.3 pA/p, P = 0.013), indicating that the sodium current was inhibited by amiloride, an ENaC inhibitor (Figure 2B).
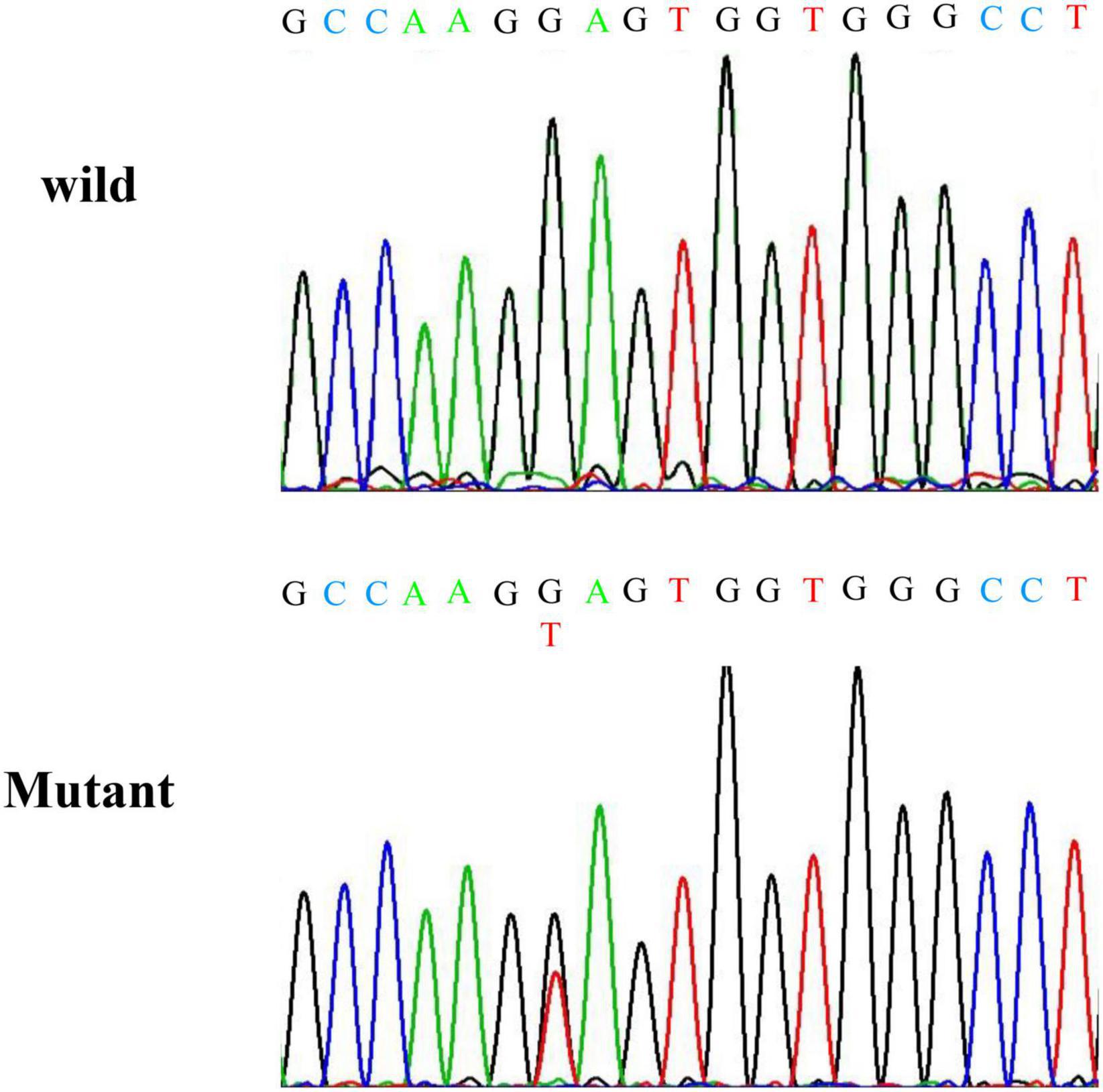
Figure 1. Results of Sanger sequencing. Sanger sequencing indicates a nonsense mutation at codon position 562 of exon 13 of SCNN1G.
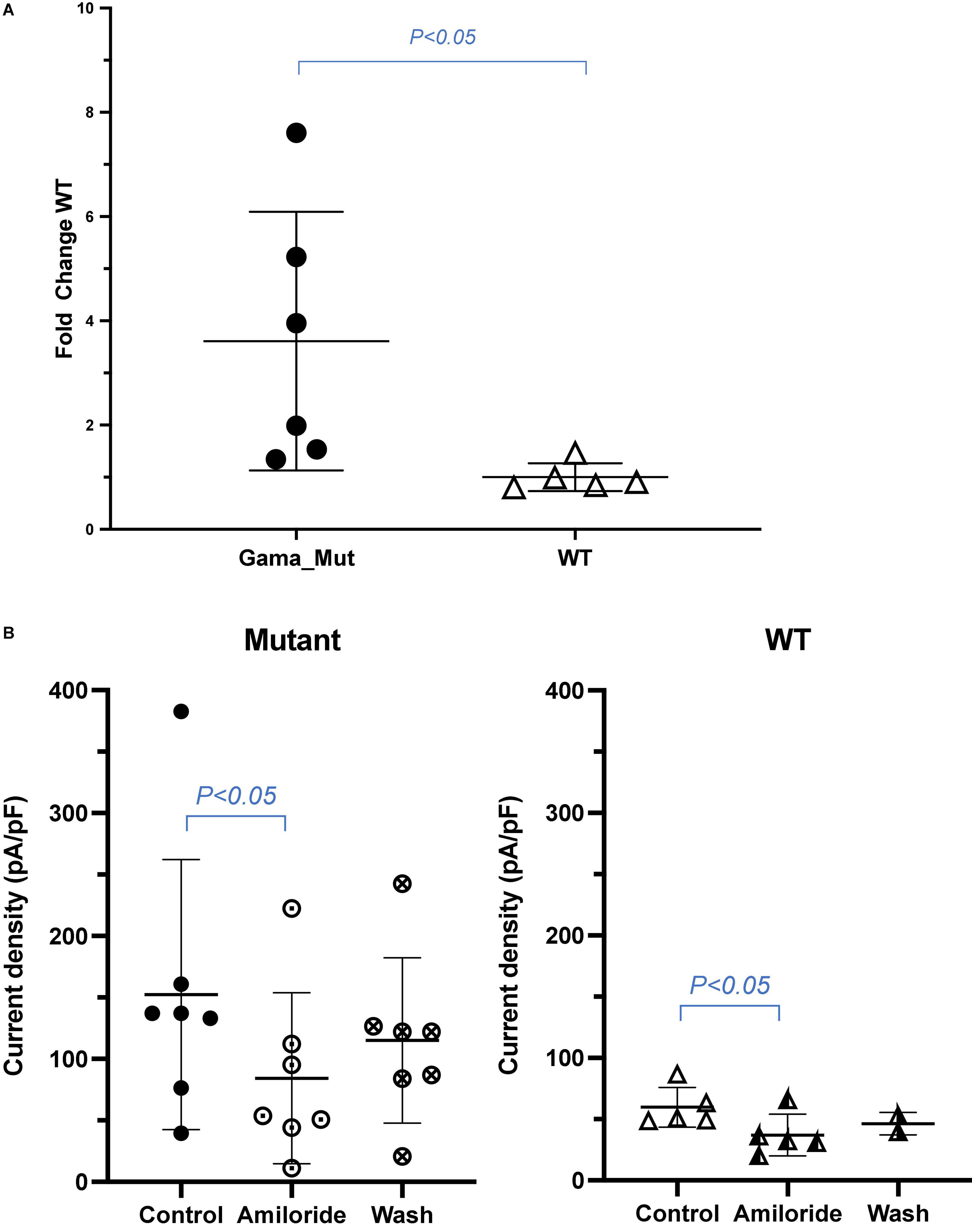
Figure 2. (A) Sodium current between wild and mutant group. Amiloride-sensitive sodium current was increased in CHO expressing mutant ENaC compared with those expressing wild type channels. Values are significantly different between wild type. (B) Amiloride inhibition of sodium current in wild and mutant group. Amiloride-sensitive sodium current was decreased in CHO of mutant ENaC after adding amiloride compared with controlled group. Values are significantly different between with amiloride and without amiloride (P < 0.05).
According to the search strategy, A total of 29 patients from 7 families, and 3 sporadic patients harbored different mutations were included (Table 2). Ten SCNN1G mutations, including 4 nonsense mutations, 3 missense mutations, and 3 frameshift mutations were found, 3 of them were validated for pathogenicity by patch-clamp monitoring of amiloride-sensitive sodium currents. Except for the patients with missing blood pressure values, all other patients were diagnosed with hypertension. Among included patients, 69% had hypokalemia, 38% had suppressed PAC, and 79% had suppressed PRA. Four patients (14%) had cerebrovascular events.
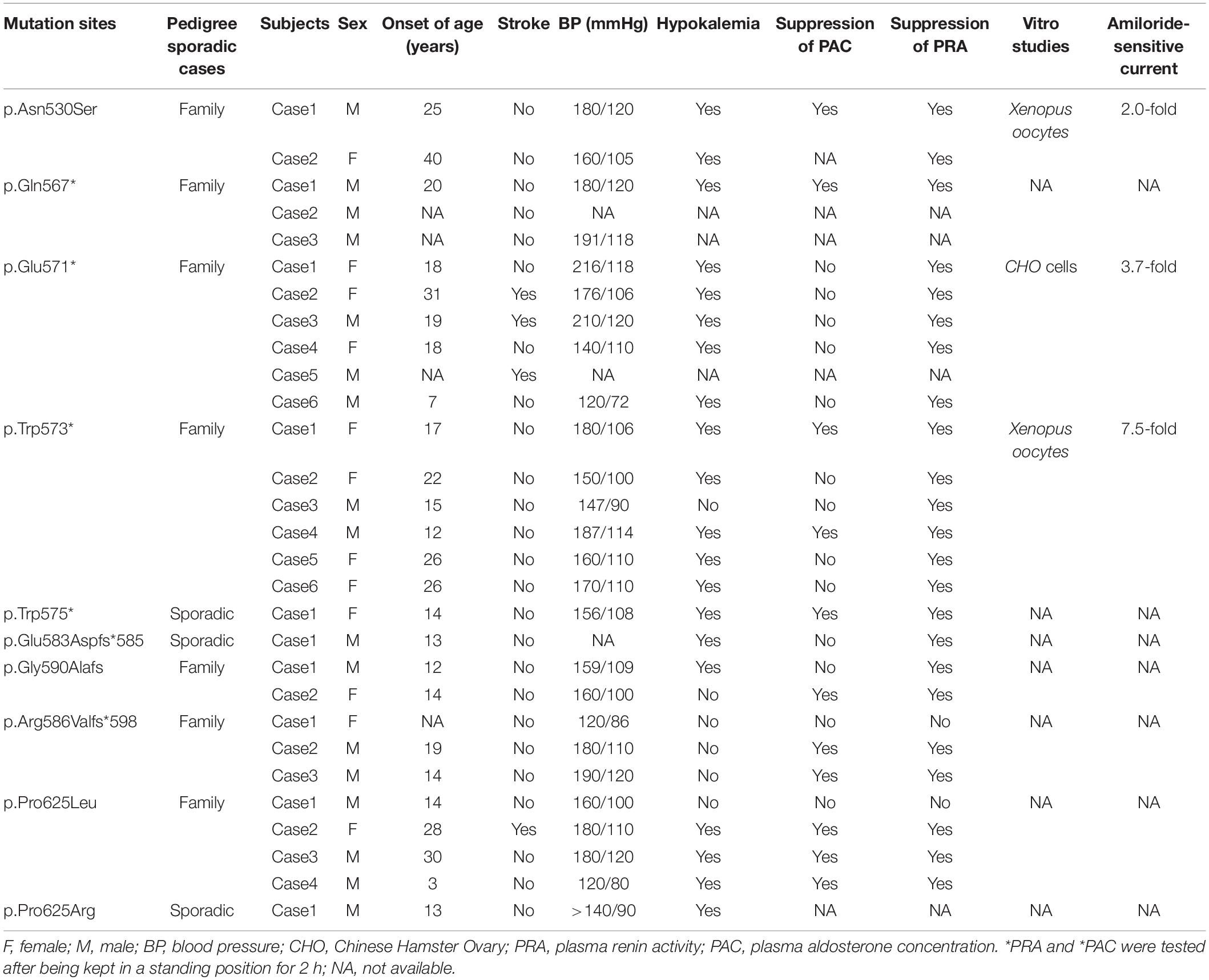
Table 2. Results of systematic review of published studies regarding LS patients carrying SCNN1G mutations.
In this study, the pathogenicity of a nonsense mutation (c.1171 G > T) in SCNN1G was confirmed by functional experiments. The blood pressure and electrolyte concentrations of LS patients in this family returned to normal after 1 month of amiloride treatment, and no target organ damage and cardiovascular events occurred over a 7-year follow-up.
LS is a rare autosomal dominant disease, the prevalence reported as 0.91 and 1.52%, respectively (2, 17). LS is caused by mutations in SCNN1A, SCNN1B, and SCNN1G, which encode ENaCs in kidney tubules, affecting sodium reabsorption (18). Proline-rich segments in the C terminals of β and γ subunits, which are critical for function, are known as PY motifs (19). The ubiquitin ligase NEDD4 binds to ENaC PY motifs, leading to the ubiquitination and degradation of ENaC (20). Mutations typically prevent the ubiquitination of subunits, thus inhibiting the rate at which they are internalized from the membrane, resulting in increased ENaC enrichment and elevated channel activity (3). However, a minority of mutations increase ENaC activity by changing the open probability in the membrane (20, 21). In 2017, a SCNN1A mutation, a gain-of-function mutation in the extracellular domain of the α subunit, was reported, which primarily increased channel open probability rather than channel surface density (8). By altering ENaC activity, sodium reabsorption increases, which leads to volume expansion and hypertension.
There are 29 mutation sites linked to LS, and nearly all mutations delete or alter PY motifs, and include frameshift, nonsense, or missense mutations (12, 20, 22–24). Among the SCNN1G genetic spectrum, 10 sites have been detected (3, 13, 22, 23), including four nonsense mutations, three missense mutations, and three frameshift mutations (Table 2). The first identified SCNN1G mutation was a nonsense mutation (p.Trp573*) reported by Hansson et al.; Xenopus oocytes expressing mutant p.Trp573* displayed 7.5-fold increase in amiloride-sensitive sodium current than wild-type (25). In the following years, a nonsense mutation site (p.Trp575*) was detected in a sporadic case with hypertension in Japan (26), and Shi et al. and Zhang et al. reported a p.Gln567* mutation in a Chinese family and sporadic case, respectively (27, 28). However, pathogenic functional experiments were not conducted for the p.Gln567* mutation. Three frame mutations and one missense site have been reported in different families, and their clinical features are summarized in Table 2. Most mutation sites impact PY motifs, except for p.Asn530Ser. The p.Asn530Ser mutation is in the extracellular domain and does not influence the PY motif, but it does cause an LS phenotype (21). In patch-clamp experiments, the p.Asn530Ser mutation presented twofold higher amiloride-sensitive currents compared with the wild type, by increasing the open probability of channels (21). The p.Glu571* mutation was first reported by our team in a Chinese family, and this variant has also been identified in another Chinese family (2, 17). In the present functional experiments, cells with the p.Glu571* mutation displayed 3.7-fold amiloride-sensitive currents of wild-type cells.
LS is a kind of channelopathy, and patch-clamp experiments are very important for verifying ion channel diseases. According to previous research, pathogenic mutations in SCNN1G mostly affect the PY motif, but pathogenicity has only been functionally verified in three sites. The physiological characteristics of ENaCs expressed in Xenopus oocytes are similar to those of ENaCs in human distal renal tubules (29); thus, functional experiments usually use Xenopus oocytes for detecting current variations in patch-clamp experiments. In the present study, however, we use CHO cells to detect current variations. CHO cells are used in patch-clamp electrophysiological experiments because of their low endogenous expression of ion channels (30). Amiloride-sensitive-sodium currents were suppressed after adding amiloride in the present study, indicating drug efficacy from an ion channel aspect. Different variants display different degrees of amiloride-induced sodium current changes, which may be associated with the effects of mutation sites on PY motifs and the cell model. Compared with extracellular domain mutations, PY motif mutations expressed higher amiloride-sensitive currents, indicating that PY motif mutations have a greater impact on ENaC activity.
Phenotypes vary greatly in patients harboring different mutations, as well as for the same mutation in a pedigree (Table 2). A systematic review conducted in 2018 reported that 92.4% of patients with LS presented hypertension (3). In the current pedigree, all members carrying the p.Glu571* variant presented hypertension, and some even had hypertensive stroke. Moreover, one of the typical features of LS is early-onset hypertension; most patients develop hypertension before 30 years. The youngest reported patients are a 10-week-old baby, who was a sporadic case, and a 2-year-old child from an LS family (9, 31). According to the largest retrospective analysis of LS patients, the average onset age of hypertension is 15.5 ± 3.3 years (7). In our research, the index case was diagnosed with hypertension at 18 years old with left ventricular concentric hypertrophy. We speculated that the proband probably had hypertension in child, and she had no obvious symptoms and had not been diagnosed in time. Another 7-year-old pediatric patient had hypertension, hypokalemia, and no target organ damage, as well he had no obvious symptoms and his onset age is unclear. The remaining family members diagnosed with hypertension are almost less than 30 years old.
In this family, there was heterogeneity not only in the occurrence of high blood pressure, but also in the severity of hypertension and complications. Reported complications of persistent hypertension in LS patients include cerebrovascular accidents, early death, renal insufficiency, nose bleeds, and retinal damage (7, 12, 32). Cerebrovascular accidents before the age of 40 were common in this LS family, especially stroke, which causes a heavy burden to patients and their families. The reason for the high incidence of stroke in this family may include poor management of blood pressure, genotype, and environmental factors.
Hypokalemia is present in 71.8% of cases of LS (3). In the current study, all mutation-carrying participants exhibited hypokalemia. Tamura et al. and Fan Peng et al. reached a similar conclusion: that some LS patients have hypertension but exhibit normokalemia (13, 33). The suppression of PRA and PAC is considered a typical presentation, and can be used to differentiate other possible causes of secondary hypertension (34). In our study, all members presented with suppression of PRA, which is in line with the typical characteristics of LS. However, the PAC levels of all members were within the normal range, and members harboring the same variant in another LS pedigree also presented normal PAC (17). The presentation of PAC shows clear heterogeneity compared with other variant-related phenotypes. The reasons for the heterogeneity of LS may be related to aspects including environmental factors and gene polymorphisms. LS patients with SCNN1A mutation may presented with mild phenotype than typical LS, which may attribute to different pathogenetic mechanisms (8). Therefore, for patients with suspected LS phenotype but milder symptoms, the possibility of carrying SCNN1A mutation should be considered.
Clinical presentations and laboratory findings in LS are heterogeneous, which can hamper its diagnosis. Family history can provide clues, and combined with early-onset hypertension and laboratory results, can be used to identify suspected LS patients. Inadequate symptoms and insufficient awareness of children with monogenic hypertension leading to fail to intervene in time, and most of them have developed complications in youth (12). According to the previous study, 90% pediatric patients had LS family history, so genetic screening can provide definitive confirmation of adult, and pediatric LS (22, 35). Additionally, investigation of the pathogenicity of variants based on amiloride-sensitive currents is needed.
There were several limitations in the present study. This site was not a novel mutation—it has been briefly mentioned in a review by our team. Furthermore, we only compared the amiloride-sensitive sodium currents of γGlu571* mutations with wild-type channels, and not with other truncation mutations of the γ subunit, which may explain the heterogeneous results to some extent. The tailored treatment with amiloride efficacy in blood pressure control and avoiding cardiovascular events has only been validated in the long-term follow-up results of this family, which cannot be representative of all LS patients. A cohort study of LS patients is needed to explore the efficacy of amiloride in improving prognosis and blood pressure control.
In conclusion, wen identified the pathogenicity of a nonsense mutation by functional analysis. This discovery not only expands the spectrum of SCNN1G mutations, but also increases the pathogenicity grades. Tailored treatment with amiloride may be an effective treatment for long-term blood pressure management and preventing cardiovascular events, so timely intervention makes a difference for adult and pediatric patients.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by the Fuwai Hospital, National Center for Cardiovascular Diseases. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
DZ, Y-XL, and X-LZ designed the study and modified the manuscript. DZ, YQ, X-QD, C-XY, and X-CL collected clinical information and performed data analysis. DZ, Y-TL, K-QY, PF, YX-H, and L-GG performed experiments. DZ wrote the manuscript. All authors reviewed this work.
This work was supported by the National Key Research and Development Program of China (2016YFC1300100), the National Natural Science Foundation of China (81974042), and the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2019XK320058 and 2019XK320057).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank all participants for taking part in this study and agencies that aided the efforts of the authors.
1. Liddle GW. A familial renal disorder stimulating primary aldosteronism but with negligible aldosterone secretion. Trans Assoc Am Physicians. (1963) 76:199–213.
2. Wang LP, Yang KQ, Jiang XJ, Wu HY, Zhang HM, Zou YB, et al. Prevalence of liddle syndrome among young hypertension patients of undetermined cause in a Chinese population. J Clin Hypertens (Greenwich). (2015) 17:902–7. doi: 10.1111/jch.12598
3. Tetti M, Monticone S, Burrello J, Matarazzo P, Veglio F, Pasini B, et al. Liddle syndrome: review of the literature and description of a new case. Int J Mol Sci. (2018) 19:812. doi: 10.3390/ijms19030812
4. Staub O, Abriel H, Plant P, Ishikawa T, Kanelis V, Saleki R, et al. Regulation of the epithelial Na+ channel by Nedd4 and ubiquitination. Kidney Int. (2000) 57:809–15. doi: 10.1046/j.1523-1755.2000.00919.x
5. Zhou R, Kabra R, Olson DR, Piper RC, Snyder PM. Hrs controls sorting of the epithelial Na+ channel between endosomal degradation and recycling pathways. J Biol Chem. (2010) 285:30523–30. doi: 10.1074/jbc.M110.150755
6. Botero-Velez M, Curtis JJ, Warnock DG. Brief report: Liddle’s syndrome revisited–a disorder of sodium reabsorption in the distal tubule. N Engl J Med. (1994) 330:178–81. doi: 10.1056/NEJM199401203300305
7. Cui Y, Tong A, Jiang J, Wang F, Li C. Liddle syndrome: clinical and genetic profiles. J Clin Hypertens (Greenwich). (2017) 19:524–9. doi: 10.1111/jch.12949
8. Salih M, Gautschi I, van Bemmelen MX, Di Benedetto M, Brooks AS, Lugtenberg D, et al. A missense mutation in the extracellular domain of alphaenac causes Liddle syndrome. J Am Soc Nephrol. (2017) 28:3291–9. doi: 10.1681/ASN.2016111163
9. Findling JW, Raff H, Hansson JH, Lifton RP. Liddle’s syndrome: prospective genetic screening and suppressed aldosterone secretion in an extended kindred. J Clin Endocrinol Metab. (1997) 82:1071–4. doi: 10.1210/jcem.82.4.3862
10. Rayner BL, Owen EP, King JA, Soule SG, Vreede H, Opie LH, et al. A new mutation, R563q, of the beta subunit of the epithelial sodium channel associated with low-renin, low-aldosterone hypertension. J Hypertens. (2003) 21:921–6. doi: 10.1097/00004872-200305000-00016
11. Fan P, Lu CX, Zhang D, Yang KQ, Lu PP, Zhang Y, et al. Liddle syndrome misdiagnosed as primary aldosteronism resulting from a novel frameshift mutation of Scnn1b. Endocr Connect. (2018) 7:1528–34. doi: 10.1530/EC-18-0484
12. Fan P, Zhang D, Pan XC, Yang KQ, Zhang QY, Lu YT, et al. Premature stroke secondary to severe hypertension results from liddle syndrome caused by a novel Scnn1b mutation. Kidney Blood Press Res. (2020) 45:603–11. doi: 10.1159/000507580
13. Fan P, Zhao YM, Zhang D, Liao Y, Yang KQ, Tian T, et al. A Novel frameshift mutation of Scnn1g causing Liddle syndrome with normokalemia. Am J Hypertens. (2019) 32:752–8. doi: 10.1093/ajh/hpz053
14. Fan P, Lu CX, Yang KQ, Lu PP, Hao SF, Luo F, et al. Truncated epithelial sodium channel beta subunit responsible for Liddle syndrome in a Chinese family. Kidney Blood Press Res. (2019) 44:942–9. doi: 10.1159/000500919
15. Flynn JT, Kaelber DC, Baker-Smith CM, Blowey D, Carroll AE, Daniels SR, et al. Clinical practice guideline for screening and management of high blood pressure in children and adolescents. Pediatrics. (2017) 140:e20171904. doi: 10.1542/peds.2017-1904
16. Ilatovskaya DV, Pavlov TS, Levchenko V, Negulyaev YA, Staruschenko AJTFJ. Cortical actin binding protein cortactin mediates enac activity via Arp2/3 complex. FASEB J. (2011) 25:2688–99. doi: 10.1096/fj.10-167262
17. Liu K, Qin F, Sun X, Zhang Y, Wang J, Wu Y, et al. Analysis of the genes involved in mendelian forms of low-renin hypertension in Chinese early-onset hypertensive patients. J Hypertens. (2018) 36:502–9. doi: 10.1097/HJH.0000000000001556
18. Rossier BC. Epithelial sodium channel (Enac) and the control of blood pressure. Curr Opin Pharmacol. (2014) 15:33–46. doi: 10.1016/j.coph.2013.11.010
19. Schild L, Lu Y, Gautschi I, Schneeberger E, Lifton RP, Rossier BC. Identification of a Py motif in the epithelial na channel subunits as a target sequence for mutations causing channel activation found in Liddle syndrome. Embo J. (1996) 15:2381–7. doi: 10.1002/j.1460-2075.1996.tb00594.x
20. Enslow BT, Stockand JD, Berman JM. Liddle’s syndrome mechanisms, diagnosis and management. Integr Blood Press Control. (2019) 12:13–22. doi: 10.2147/IBPC.S188869
21. Hiltunen TP, Hannila-Handelberg T, Petajaniemi N, Kantola I, Tikkanen I, Virtamo J, et al. Liddle’s syndrome associated with a point mutation in the extracellular domain of the epithelial sodium channel gamma subunit. J Hypertens. (2002) 20:2383–90. doi: 10.1097/01.hjh.0000042881.24999.fc
22. Fan P, Pan XC, Zhang D, Yang KQ, Zhang Y, Tian T, et al. Pediatric Liddle syndrome caused by a novel Scnn1g variant in a chinese family and characterized by early-onset hypertension. Am J Hypertens. (2020) 33:670–5. doi: 10.1093/ajh/hpaa037
23. Lata S, Marasa M, Li Y, Fasel DA, Groopman E, Jobanputra V, et al. Whole-exome sequencing in adults with chronic kidney disease: a pilot study. Ann Intern Med. (2018) 168:100–9. doi: 10.7326/M17-1319
24. Brower RK, Ghlichloo IA, Shabgahi V, Elsholz D, Menon RK, Vyas AK. Liddle syndrome due to a novel C.1713 deletion in the epithelial sodium channel beta-subunit in a normotensive adolescent. AACE Clin Case Rep. (2021) 7:65–8. doi: 10.1016/j.aace.2020.11.017
25. Hansson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Lu Y, et al. Hypertension caused by a truncated epithelial sodium channel gamma subunit: genetic heterogeneity of Liddle syndrome. Nat Genet. (1995) 11:76–82. doi: 10.1038/ng0995-76
26. Yamashita Y, Koga M, Takeda Y, Enomoto N, Uchida S, Hashimoto K, et al. Two sporadic cases of Liddle’s syndrome caused by de novo enac mutations. Am J Kidney Dis. (2001) 37:499–504. doi: 10.1053/ajkd.2001.22072
27. Shi JY, Chen X, Ren Y, Long Y, Tian HM. [Liddle’s syndrome caused by a novel mutation of the gamma-subunit of epithelial sodium channel gene Scnn1g in Chinese]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. (2010) 27:132–5. doi: 10.3760/cma.j.issn.1003-9406.2010.02.003
28. Zhang P, Kong LQ, Huang DJ. [Liddle syndrome complicating with nonfunctional adrenal cortical adenoma: a case report]. Zhonghua Xin Xue Guan Bing Za Zhi. (2017) 45:331–2. doi: 10.3760/cma.j.issn.0253-3758.2017.04.015
29. Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, et al. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. (1994) 367:463–7. doi: 10.1038/367463a0
30. Gamper N, Stockand JD, Shapiro MS. The use of Chinese hamster ovary (Cho) cells in the study of ion channels. J Pharmacol Toxicol Methods. (2005) 51:177–85. doi: 10.1016/j.vascn.2004.08.008
31. Assadi FK, Kimura RE, Subramanian U, Patel S. Liddle syndrome in a newborn infant. Pediatr Nephrol. (2002) 17:609–11. doi: 10.1007/s00467-002-0897-z
32. Gong L, Chen J, Shao L, Song W, Hui R, Wang Y. Phenotype-genotype analysis in two Chinese families with Liddle syndrome. Mol Biol Rep. (2014) 41:1569–75. doi: 10.1007/s11033-013-3003-7
33. Tamura H, Schild L, Enomoto N, Matsui N, Marumo F, Rossier BC. Liddle disease caused by a missense mutation of beta subunit of the epithelial sodium channel gene. J Clin Invest. (1996) 97:1780–4. doi: 10.1172/JCI118606
34. Mumford E, Unwin RJ, Walsh SB. Liquorice, Liddle, Bartter or Gitelman-how to differentiate? Nephrol Dial Transplant. (2019) 34:38–9. doi: 10.1093/ndt/gfy199
Keywords: Liddle syndrome, pathogenicity, pediatrics, amiloride-sensitive current, longterm prognosis
Citation: Zhang D, Qu Y, Dong X-Q, Lu Y-T, Yang K-Q, Liu X-C, Fan P, Hu Y-X, Yang C-X, Gao L-G, Liu Y-X and Zhou X-L (2022) Pathogenicity and Long-Term Outcomes of Liddle Syndrome Caused by a Nonsense Mutation of SCNN1G in a Chinese Family. Front. Pediatr. 10:887214. doi: 10.3389/fped.2022.887214
Received: 01 March 2022; Accepted: 02 May 2022;
Published: 24 May 2022.
Edited by:
Naglaa M. Kamal, Cairo University, EgyptReviewed by:
Vandana Jain, All India Institute of Medical Sciences, IndiaCopyright © 2022 Zhang, Qu, Dong, Lu, Yang, Liu, Fan, Hu, Yang, Gao, Liu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ya-Xin Liu, eWF4aW5saXUxOTc4QGhvdG1haWwuY29t; Xian-Liang Zhou, emhvdXhpYW5saWFuZzAzMjZAaG90bWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.