 Soon-Min Hong
Soon-Min Hong Wei Chen
Wei Chen Jiaqi Feng
Jiaqi Feng Dai Dai
Dai Dai Nan Shen
Nan Shen- 1Shanghai Institute of Rheumatology, School of Medicine, Renji Hospital, Shanghai Jiao Tong University, Shanghai, China
- 2Department of Rheumatology, School of Medicine, Renji Hospital, Shanghai Jiao Tong University, Shanghai, China
- 3Department of Pediatrics, School of Medicine, Renji Hospital, Shanghai Jiao Tong University, Shanghai, China
- 4Shenzhen Futian Hospital for Rheumatic Diseases, Shenzhen, China
- 5State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, School of Medicine, Renji Hospital, Shanghai Jiao Tong University, Shanghai, China
- 6Center for Autoimmune Genomics and Etiology (CAGE) and Divisions of Rheumatology, Cincinnati Children's Hospital Medical Center, Cincinnati, OH, United States
- 7Joint Research Laboratory for Rheumatology of Shenzhen University Health Science Center and Shenzhen Futian Hospital for Rheumatic Diseases, Shenzhen, China
Background: The study of genetic predisposition to pediatric systemic lupus erythematosus (pSLE) has brought new insights into the pathophysiology of SLE, as it is hypothesized that genetic predisposition is greater in children. Furthermore, identifying genetic variants and linking disrupted genes to abnormal immune pathways and clinical manifestations can be beneficial for both diagnosis and treatment. Here, we identified genetic alterations in a patient with childhood-onset SLE and analyzed the immunological mechanisms behind them to support future diagnosis, prognosis, and treatment.
Methods: Whole exome sequencing (WES) was adopted for genetic analysis of a patient with childhood-onset SLE. Gene mutations were confirmed by Sanger sequencing. Clinical data of this patient were collected and summarized. Ingenuity Pathway Analysis was used to provide interacting genes of the perturbed genes. Online Enrichr tool and Cytoscape software were used to analysis the related pathways of these genes.
Results: We present a case of a 2-year-old girl who was diagnosed with idiopathic thrombocytopenic purpura (ITP) and SLE. The patient was characterized by cutaneous bleeding spots on both lower extremities, thrombocytopenia, decreased serum complements levels, increased urinary red blood cells, and positive ANA and dsDNA. The patient was treated with methylprednisolone and mycophenolate, but clinical remission could not be achieved. The genomic analysis identified three novel mutations in this pSLE patient, a double-stranded missense mutation in ACP5 (c.1152G>T and c.420G>A) and a single-stranded mutation in SAMHD1 (c.1423G>A). Bioinformatic analysis showed that these two genes and their interacting genes are enriched in the regulation of multiple immune pathways associated with SLE, including cytokine signaling and immune cell activation or function. Analysis of the synergistic regulation of these two genes suggests that abnormalities in the type I interferon pathway caused by genetic variants may contribute to the pathogenesis of SLE.
Conclusion: The combined complexity of polymorphisms in the coding regions of ACP5 and SAMHD1 influences the susceptibility to SLE. Alterations in these genes may lead to abnormalities in the type I interferon pathway. Our study extends the spectrum of mutations in the ACP5 and SAMHD1 genes. The identification of these mutations could aid in the diagnosis of SLE with genetic counseling and suggest potential precise treatments for specific pathways.
Introduction
Systemic lupus erythematosus (SLE) is a chronic multisystem autoimmune disease characterized by the production of autoantibodies and overactivation of the type I interferon (IFN) pathway, with alternating exacerbations and remissions (1). SLE is a heterogeneous disease and the diagnosis of SLE requires the presence of 4 of the 11 defining criteria. Clinical manifestations of SLE patients include arthritis, rash, serositis, various cytopenias, renal disease, psychiatric, neurological, and other manifestations (2, 3). Despite the wide variability, approximately 50% of SLE patients have persistently elevated blood levels of type I IFN over time. In addition, 60–80% of SLE patients have increased expression of interferon-stimulated genes (ISG) in peripheral blood, which is referred to as an IFN signature (1, 4). As with other heterogeneous diseases, the pathogenesis of SLE remains unclear. However, heritability has long been recognized as an important causative factor, affecting approximately 66% in SLE (5). To date, genome-wide association studies (GWAS) have identified more than 80 loci closely associated with lupus, and single-gene susceptibility to SLE has been suggested to be caused by single-nucleotide mutations in the coding regions of nearly 30 genes (6–8). The current view linking many susceptibility genes to key immune pathways is consistent with previous experimental studies including the role of immune complexes, host immune signaling, and interferon pathways in the pathogenesis of SLE (8, 9). Monogenic type I interferon diseases such as spinal chondrodystrophy (SPENCD) and Aicardi-Goutières syndrome (AGS) clinically overlap with SLE, both of which are associated with spontaneous type I IFN responses with no detectable exogenous viral infection (10). SPENCD is driven by an autosomal recessive mutation in ACP5. AGS can have different patterns of inheritance, mostly caused by single mutations in the ADAR, TREX1, RNASEH2A, RNASEH2B, RNASEH2C or SAMHD1 genes (11). These studies, together with laboratory observations, suggest that IFN plays an important role in the pathogenesis of SLE.
Given the complexity and heterogeneity of SLE, identifying genetic alterations and linking them to underlying immunologic pathogenesis could improve individualized diagnostic and therapeutic approaches to SLE. Here, we identified a pediatric-onset SLE patient with germline mutations in both ACP5 and SAMHD1.This patient carried three novel mutations from both father and mother, resulting in a double missense mutation in ACP5 (c.1152G>T in the mother and c.420G>A in the father) and a single missense mutation in SAMHD1 (c.1423G>A in the mother). We found that these two genes and their regulatory networks were enriched in multiple immune pathways associated with SLE, including cytokine signaling and immune cell activation or function. We also analyzed the synergistic regulation of these two genes and demonstrated their combined contribution to type I IFN, which suggests a pathogenic role of this genetic variant in this pSLE patient.
Materials and Methods
Human Patients
We collected and summarized the clinical data of a patient who was diagnosed with lupus and carried specific gene mutations. Laboratory results before treatment were recorded for analysis. Written informed consent to participatein this study was provided by the participants' legal guardian/next of kin. Protocol of this study was approved by the Renji Hospital Biobank is funded by the National Human Genetic Resources Sharing Service Platform (2005DKA21300).
DNA Sequencing
DNA from probands and their family members were isolated and purified from blood and prepared for whole-exome sequencing (WES). DNA samples were enriched with Human SureSelect XT2 All Exon V4 Kit and sequenced by Illumina HiSeq 2000 (Illumina, Inc.). WES had 21% low or uncovered exon bases. Bioinformatic analysis was performed at JCSMR, ANU. Raw sequence reads were aligned to the reference genome (Hg19) and single-nucleotide variants and small insertions and deletions called using GATK. All SNVs of interest in ACP5 and SAMHD1 were confirmed by Sanger sequencing. Amplifluor to detect ACP5G290V, ACP5R46Q, and SAMHD1R408H in the APOSLE cohort was performed using the CHEMICON Amplifluor SNPs HT Genotyping System Fam-Joe kit S7909 (Merck-Millipore). The 45 and Up (12, 13) and ASPREE (14) datasets were used as reference healthy controls, accessed through the MGRB Collaborative (http://sgc.garvan.org.au/mgrb/initiatives).
WES Data Processing and Batch Correction
Probes were filtered out if the detection P value was greater than 0.01 for at least 100% of the samples. All data values <10 was set to 10 and then the data were log2 transformed. An additional filter selecting the 75% most variable transcripts was performed, leaving a total 18,004 probes for analysis. Principal variance component analysis (PVCA) was conducted to identify undesirable sources of technical variability within the data and batch correction was applied to correct for this technical variation. Both PVCA and batch correction were conducted using JMP Genomics 7.0 (SAS Institute) analysis software.
Protein Preparation and Mutations Interactions Analysis
The X-ray resolved crystal structures of wild type ACP5 (PDB: 2BQ8) and SAMHD1 (PDB: 7A5Y) were download from RCSB PDB database, the mutations' structures were generated by pymol2.2. Water molecules, ions, heteroatoms, and all ligands were eliminated. Finally, the protonation states of the wild and mutated structures were then deliberated using a H++ server. Additionally, all missing hydrogen atoms were inserted by pymol2.2 The interactions analysis was used pymol2.2.
Statistical and Bioinformatics Analysis With IPA, Cytoscape
We used BioProfiler searches the Ingenuity Pathway Analysis (IPA, Qiagen) database to explore associations gene and phenotypes. IPA was used to construct pathways and networks (15, 16), to quickly identify biological relationships, mechanisms, pathways, functions and diseases most relevant to experimental datasets. Then we obtain two comprehensive lists of proteins implicated in either the ACP5 or SAMHD1 by IPA. Detailed information of the expressed proteins and endogenous biochemical compounds that have been associated with genes by using the “Annotation” module in IPA. The protein-protein interaction (PPI) network based on ACP5 and SAMHD1 was obtained via String (V.11.5) to predicting the protein interactions and obtaining protein connection scores, and the interaction scores beyond 0.400 were uploaded to Cytoscape for visualization and analysis of biological networks (17, 18). Protein overlapping network identification by using CentiScaPe to investigate node centrality in both networks, and important module in the protein network related to the hub genes were abstracted by using Molecular Complex Detection (MCODE) (19). The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis from ClueGO were used to perform biological pathway enrichment analysis for the core cluster identified by MCODE (20).
Results
Case Presentation
The patient was a 2-year-old girl who had recurrent upper respiratory tract infections and oral thrush since birth. She initially presented with cutaneous bleeding spots on both lower extremities, mainly on the calves and dorsum of the feet (Figure 1A). There was no fever, cough, sputum, joint pain, or other signs of discomfort. She was seen at a local pediatric outpatient clinic, which showed thrombocytopenia (4 × 109/L) and decreased hemoglobin (98 g/L). The renal function report showed decreased blood creatinine (Bcr) level (19.3 μmol/L), urine red blood cells (373.4/ul) and urine protein (2+). Serum complement levels were decreased (C3 387 mg/L, C4 44 mg/L). No signs of hematological malignancy were found in bone marrow smear and bone marrow biopsy sections. Systemic lupus erythematosus and idiopathic thrombocytopenic purpura (ITP) were diagnosed based on skin symptoms, thrombocytopenia and decreased serum complement levels. She was then started on methylprednisolone (MP). However, the abnormalities in urine red blood cells and serum complement levels persisted.
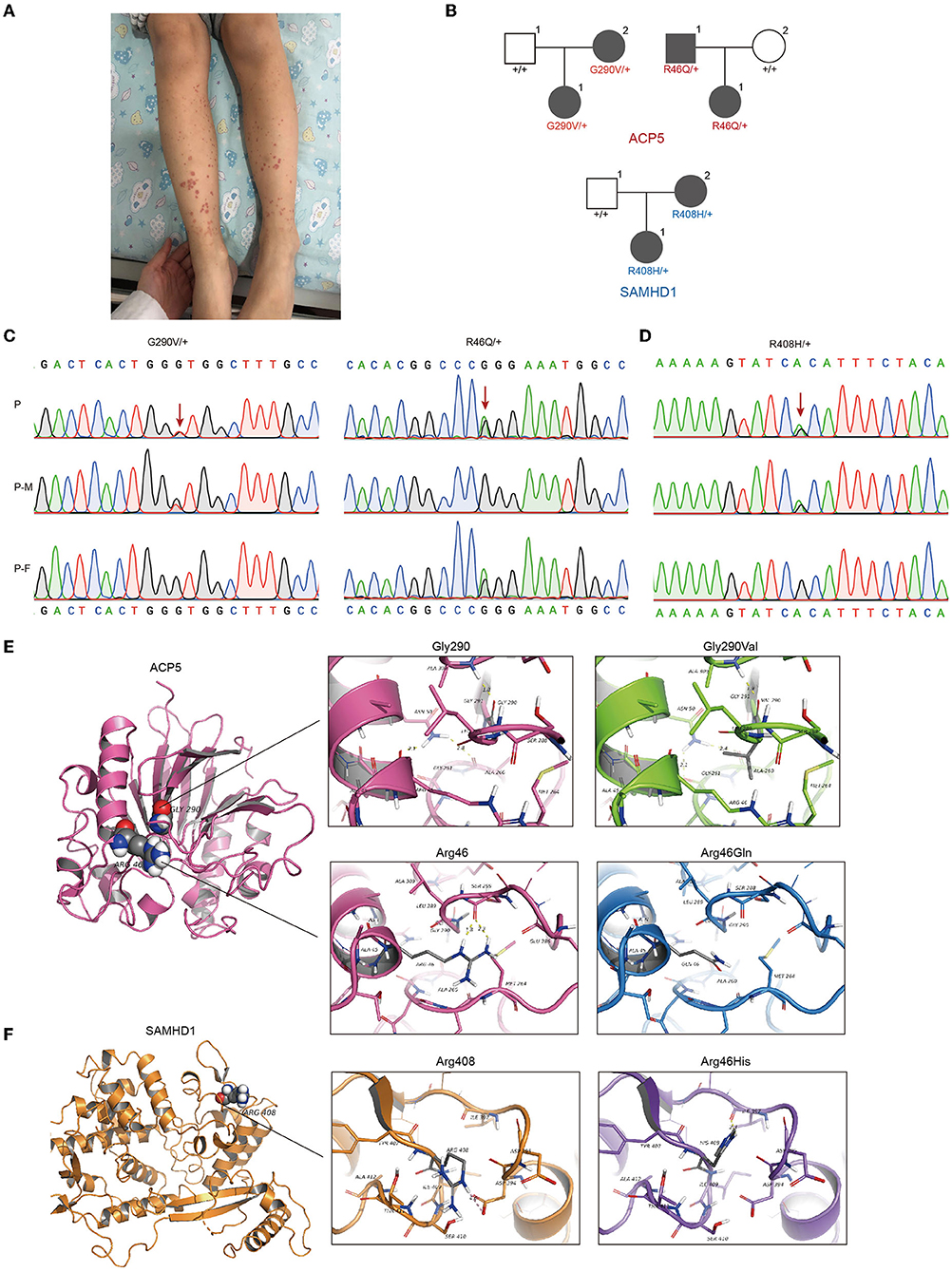
Figure 1. Identification of gene mutations in patient with pSLE. (A)The patient initially presented skin bleeding spots in both lower limbs. (B) Pedigrees showing affected probands and parents. Sanger sequencing confirmation of the c.1152G>T (p.Gly290Val), c.420G>A (p. Arg46Gln) at ACP5 (C) and c.1423G>A (p.Arg408His) at SAMHD1 (D) variant identified in our patient using exome sequencing. Chromatograms represent the reference (top) and the mutant sequence. The variant is indicated by the red arrow. (E) Top: Structure modeling of wild type and p.Gly290Val mutation of ACP5. Top left: Gly290 form H-bond with Ala309; Asn50 form H-bonds with Arg46 (Distance: 2.1Å) and Ala 260 (Distance: 2.8 Å). Top right: The mutant model shows the change of H-bonds, Asn50 form H-bonds with Gly261 (Distance: 2.1 Å) and Ala 260 (Distance: 2.4 Å). Bottom: Wild type and p.Arg46Gln mutation of ACP5. Bottom left: Arg46 form H-bonds with Ser288 (Distance: 1.8 Å and 2.3 Å). Bottom right: The mutant shows the loss of two H-bonds, this might lose the ability to form any polar interaction with other residues. (F) Wild type and p.Arg408His mutation of SAMHD1. Left: Arg408 form salt bridge with Asp394. Right: The mutant shows the loss of salt bridge with Asp394, and form H-bond with Ile397 (Distance: 2.3 Å). P, patient; P-M, mother; P-F, father.
Three months later, she presented to our pediatric clinic. Laboratory tests showed elevated white blood cell count (14.63 × 109/L), lymphocyte count (6.45 × 109/L), monocyte count (1.6 × 109/L), calcitonin (0.28 ng/ml), and decreased Bcr (16 μmol/L). Anti-nuclear antibody (ANA, >1/80), anti-ribosomal antibody (ANuA, >1) and anti-dsDNA (31.79 IU/ml) were positive. Serum complement (C3 0.66 g/L, C4 0.06 g/L), immunoglobulin (Ig) A (0.31 g/L) was decreased and IgE (308.47 IU/ml) was increased. The percentage of T lymphocytes (CD3+ 35.61%, CD4+ 19.78%, CD8+ 14.73%) in PBMC was decreased and B lymphocytes were increased (CD19+% 57.46%). Mycoplasma pneumoniae IgM antibodies were positive (1:80). Erythrocyte sedimentation rate (ESR), C-reactive protein (CRP) and renal function were normal. Cytomegalovirus (CMV) and Epstein-Barr virus (EBV) antibodies were negative. Rheumatoid factor, IgG, IgM and urine protein were all negative. Antibodies to chlamydial pneumonia, tuberculosis, TORCH, and anti-streptozotocin O were negative. Her abdominal ultrasound scan, skeletal radiography, EEG and brain MRI were normal. Immune markers such as serum immunoglobulin, complement and lymphocyte subpopulation values are listed in Table 1.
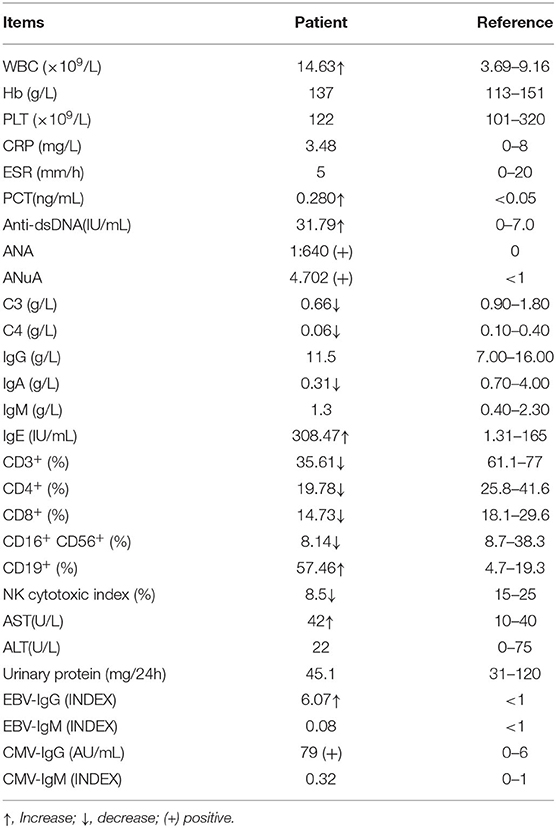
Table 1. Immune indices of patient.
Therefore, she met the diagnostic criteria for SLE and growth retardation (weight: 10.5 kg, −1.20 standard deviation (SD); height: 80 cm, −2.20 SD). She was then started on MP and mycophenolate mofetil (MMF) for 1 year and measures associated with SLE activity remained abnormal (increased dsDNA, positive ANA). At the same time, we found that the patient's IFN-α (31.56 pg/ml), IFN-γ (10.92 pg/ml), TNF-α (8.93 pg/ml), and IL-17A (23.78 pg/ml) increased dramatically during the active phase. Therefore, methotrexate (MTX) was given, but she developed leukopenia (2.76 × 109/L). Considering the inhibitory effect of MTX on bone marrow hematopoiesis, MTX was discontinued at that time and the regimen of MP, HCQ and MMF was changed. However, the patient's indicators such as dsDNA, serum complement and Ig remained unstable.
Given the early onset, multi-organ involvement and the refractory nature of alternating episodes and remissions, we should further explore the etiology to improve the clinical diagnosis and treatment of this patient.
The Germline Rare Mutations of ACP5 and SAMHD1 Identified in This Patient
Next-generation sequencing brings great convenience for accurate diagnosis. High-throughput sequencing can detect genetic alterations in an entire genome, an exome, or a group of genes. Patients with early-onset lupus often carry a high frequency of pathogenic variants, highlighting the importance of genetic testing for pSLE (21, 22). We then applied WES and identified three new mutations in this patient, all inherited from her father and mother. This patient carried a bilateral missense mutation in the ACP5 gene (NM_001111034.2) at 2 different loci (c.1152G>T [p. Gly290Val, G290V]; c.420G>A [p. Arg46Gln, R46Q]) and a unilateral missense mutation in the SAMDH1 gene (NM_15474.3) (c.1423G>A [p.ARG408HIS, R408H]). The combined annotation-dependent depletion (CADD) scores representing the deleteriousness of these three mutations were 21 (ACP5, c.1152G>T), 22 (ACP5, c.420G>A) and 6.649 (SAMDH1, c.1423G>A) (Figures 1B–D). The sites of these mutations have not been reported previously. Protein structure predictions showed that the G290V mutation resulted in weaker hydrogen bond (H bond) strength, the R46Q mutation in ACP5 resulted in the loss of two H bonds (Figure 1E), and the R408H mutation in SAMHD1 resulted in the loss of the salt bridge and the formation of an H bond (Figure 1F). We know that protein structure is closely related to function, and changes in protein structure caused by mutations may lead to disruption of biological function. Therefore, in combination with the high CADD score and altered protein structure, mutations in ACP5 may have greater pathogenicity.
Mutations in ACP5 and SAMHD1 are known to drive the pathogenesis of SPENCD and AGS, both of which are monogenic interferon diseases characterized by increased type I IFN signaling leading to vasculopathy, autoinflammation, and SLE-like disease (8, 23, 24). To date, 30 cases of SPENCD with SLE have been reported in the literature (23, 25–35) (Figure 2A; Table 2), and 9 cases of SLE with SAMHD1 mutations have been reported in the literature (21, 36–41) (Figure 2B; Table 3), including the patients in this article. Patients with SPENCD or AGS with SLE have a homogeneous clinical presentation, including autoantibody positivity, hematologic involvement, and neurologic symptomatic manifestations. Interestingly, this patient did not show clinical manifestations of epiphyseal lesions of SPENCD and neurological dysfunction of AGS in addition to autoimmune disease, implying that complex protein functional alterations due to polymorphisms in the ACP5 and SAMHD1 genes lead to different clinical symptoms. Here, we used high-throughput sequencing to identify three novel mutations in the coding regions of ACP5 and SAMHD1 in this patient that have not been previously reported to be associated with SLE.
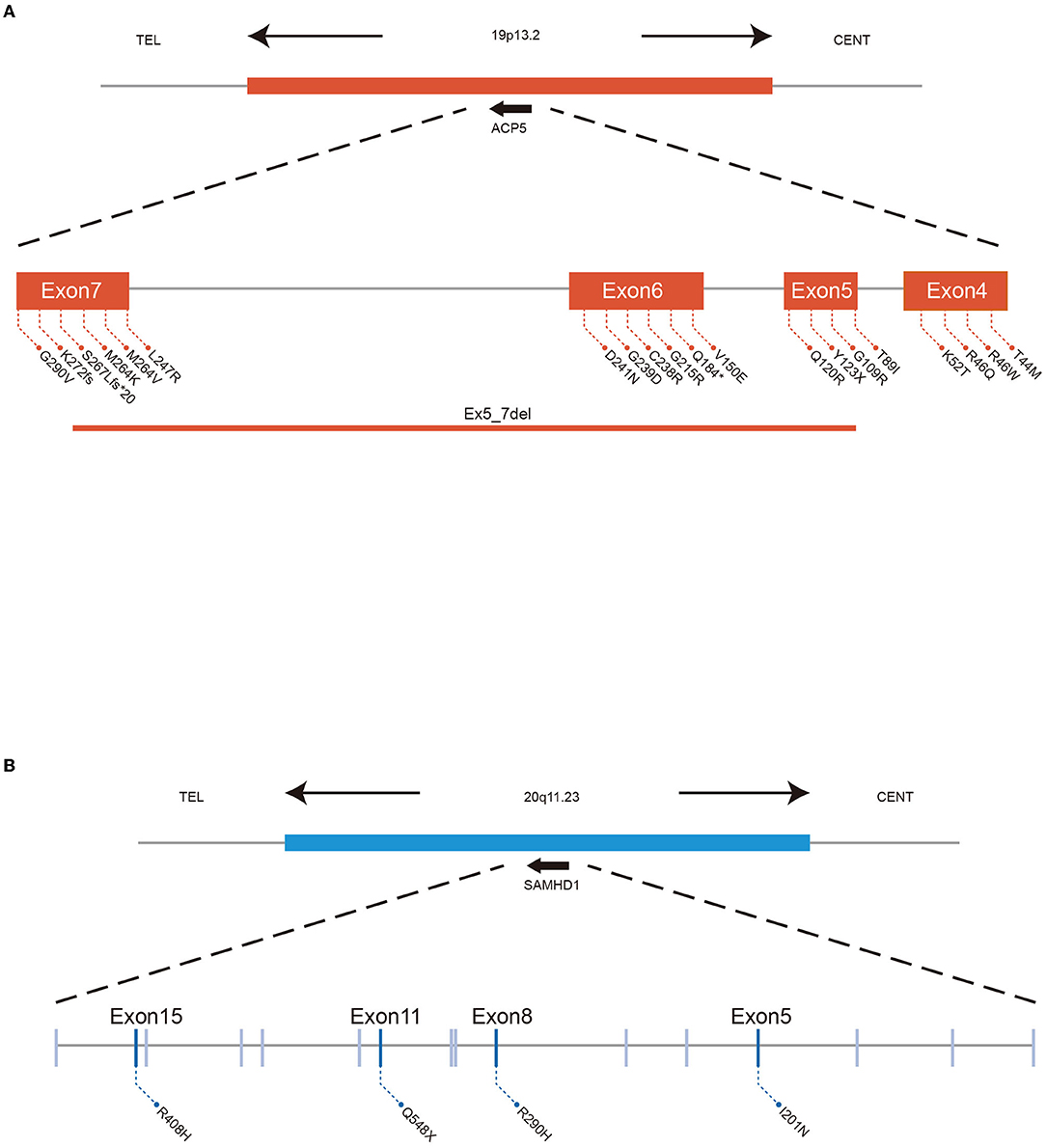
Figure 2. ACP5 mutations cause SPENCD and SAMHD1 mutations cause AGS both diseases clinically overlap with SLE, and both are associated with a spontaneous type I IFN response. (A) Summary of ACP5 mutation associated with SLE data. (B) Summary of SAMHD1 mutation associated with SLE data.
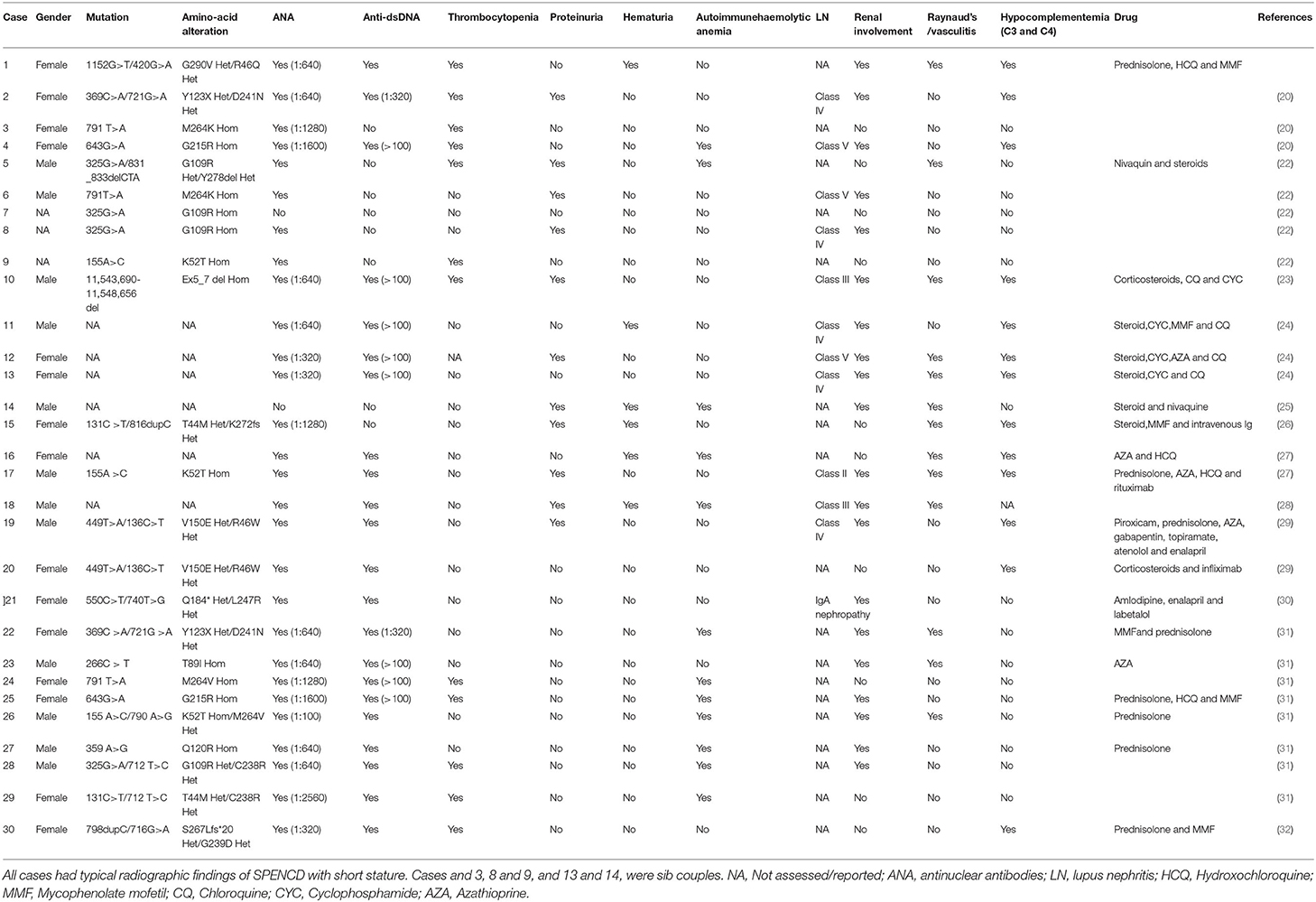
Table 2. Cases of SPENCD associated with SLE.
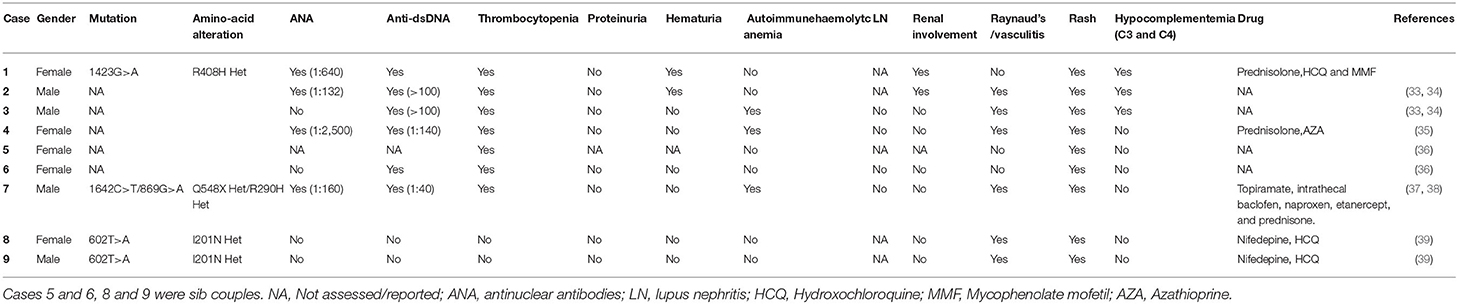
Table 3. Cases of SLE associated with mutation of SAMHD1.
The Potential Regulation Networks of ACP5 and SAMHD1 in SLE
To comprehensively summarize these two genes and their potential roles in disease, we used the IPA BioProfiler to explore the phenotype-gene relationships of ACP5 and SAMHD1 (Figure 3A). Indeed, ACP5 is associated with skeletal disorders and other diseases such as neoplastic disorders, including hematological malignancies. The same is true for SAMHD1 with inflammatory, neurological and neoplastic disorders. To further determine the regulatory roles of ACP5 and SAMHD1, we searched the interaction network of these two genes by IPA. We annotated the genome in the network by the online Enrihr tool (Figure 3B). Pathway analysis showed that ACP5 and its interacting genes are associated with osteoclast differentiation, associated with bone disorders in SPENCD. Importantly, many immune pathways were enriched, such as the Toll-like receptor signaling pathway and IL-17 signaling pathway, suggesting a potential regulatory role of ACP5 in SLE, except for type I IFN. For SAMHD1, the enriched pathways mainly were associated with viral infections, implying a dominant effect of genetic alterations in SAMHD1 on infectious diseases. SAMHD1 and its interaction network were associated with the production of type I IFN, as multiple nuclease pathways were significantly associated with it. In conclusion, we found that ACP5 and SAMHD1 are associated with multiple immune pathways that have been shown to be critical in the pathogenesis of SLE, suggesting that genetic perturbations in these two genes may lead to immunological abnormalities in these pathways.
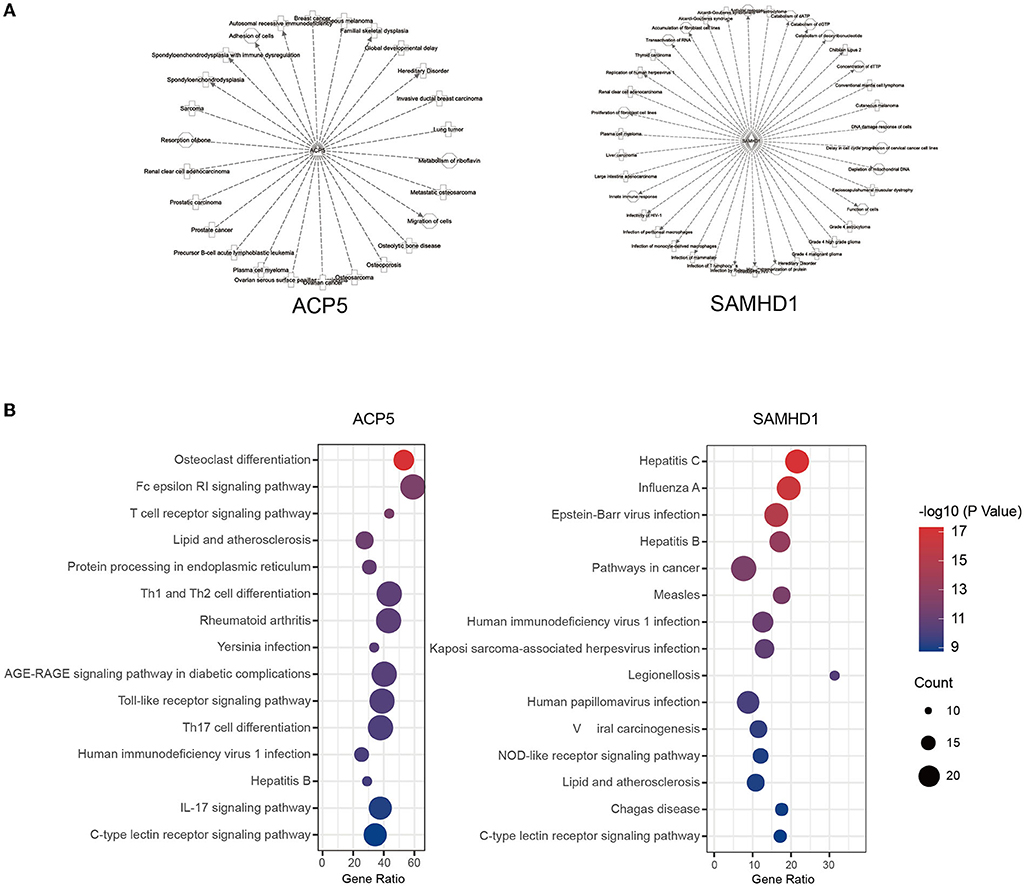
Figure 3. The potential regulation of ACP5 and SAMHD1 in SLE. The phenotype-gene relationship (A) and pathway analysis (B) were shown to be associated with immunity responses.
Interaction of ACP5 and SAMHD1 Is Associated With Interferon Pathway
Since ACP5 and SAMHD1 are associated with the type I IFN pathway, which exhibits abnormalities in both SPENCD and AGS. We interrogated the synergistic regulation of these genes by PPI network and pathway analysis and explored whether there is a link between ACP5 and SAMHD1. We constructed two protein interaction networks in Cytoscape to explore the link between ACP5 and SAMHD1 and the potential connection to the etiology of SLE (Figure 4A). The ACP5 protein interaction network included 60 nodes and 277 edges, while the SAMHD1 protein interaction network included 144 nodes and 982 edges. We merged these two networks, with blue representing nodes in the ACP5 protein interaction network and green representing nodes in the SAMHD1 protein interaction network (orange representing nodes in the duplication network). These results demonstrate a significant relationship between these genes (P-value for PPI enrichment: <1.0e-16).
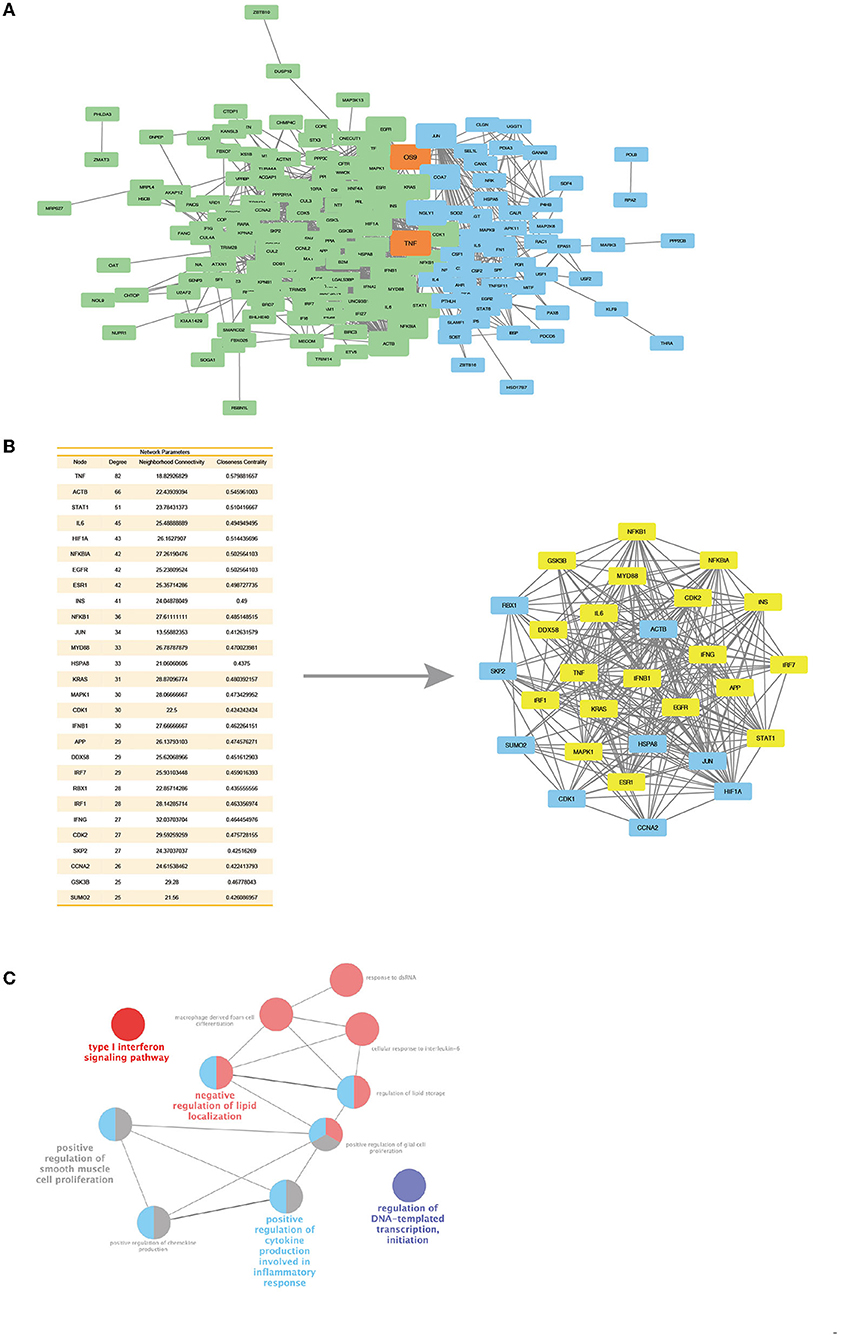
Figure 4. Protein interaction networks of ACP5 and SAMHD1 have connections and a core cluster with significant connections with two networks. (A) Merge of ACP5 protein network (green) and SAMHD1 protein network (blue) with overlap nodes in both networks (orange). The size of nodes represents the degree and betweenness of nodes. (B) Left: The table outlines the degree, neighborhood connectivity, and closeness centrality values of the core cluster nodes. Right: Networks of core cluster in merge of ACP5 protein network and SAMHD1 protein network. The yellow nodes are the densely connected MCOD clusters, based on their parameters, including connectivity, degree, and centrality. (C) ClueGo analysis identifies KEGG pathways and biological processes linked to the essential proteins in the MCOD clusters.
Next, we evaluated the central properties of the nodes in the PPI network. Highly significant nodes (score >25) had more interaction partners than non-central nodes, representing proteins with higher relevance in linking regulatory molecules. A unique protein interaction network for ACP5 and SAMHD1 was extracted by overlapping networks, including 28 nodes and 241 edges (Figure 4B). This new network was statistically significant for protein association (PPI-enriched P-value: 2.26e-11), showing highly connected edges of both networks, suggesting the possible existence of common pathways involved in the pathogenesis of SLE. To scrutinize the interconnected core clusters of the two protein interaction networks, we identified densely connected MCOD clusters and found an overlap between the ACP5 and SAMHD1 protein networks. The proteins in the core MCOD cluster include IFN-related genes, including MYD88, IRF1, IRF7, and NFKB1. These clusters were also analyzed by KEGG pathway enrichment, including the type I interferon signaling pathway. Collectively, these results further suggest that ACP5 and SAMHD1 can synergistically regulate the type I IFN pathway and that genetic alteration in these two genes accumulate susceptibility to SLE through dysregulation of the IFN pathway (Figure 4C).
Discussion
In this study, we describe the clinical manifestations of SLE in childhood, with the involvement of organs mainly affecting the skin, kidneys, and hematological system. However, several indicators of SLE remained abnormal after the patient underwent a thorough examination and treatment. We highly suspected that the patient might have some inherited genetic mutations. Then by genome sequencing, we found that the patient had novel missense mutations c.1152G>T and c.420G>A in ACP5 and c.1423G>A in SAMHD1, which were highly predictive of impaired gene function. The further bioinformatic analysis confirmed the regulatory role of these two genes in the type I IFN pathway and revealed that other immune pathways might be affected by mutations in ACP5 and SAMHD1 genes (Figure 5).
Figure 5. WES may help to analyze the potential pathogenesis of patients. Our findings suggest that the combinatorial complexity of polymorphisms in ACP5 and SAMHD1 coding regions impacts SLE susceptibility, and may contribute to the abnormal immune pathway in SLE. These mutations should be considered for potential precision therapy targeting specific pathways.
Mutations in ACP5 and SAMHD1 genes contribute to the pathogenesis of SPENCD and AGS. Both diseases clinically overlap with SLE and are associated with a spontaneous IFN response (23, 24). However, our patient did not exhibit the typical symptoms of AGS and SPENCD, neither skeletal dysplasia nor neurological dysfunction. Therefore, based on her clinical features, we diagnosed childhood-onset SLE. In addition, we observed a dramatic increase in serum IFN-α during disease flares. The patient presented with refractory SLE, with alternating exacerbations and remissions, and suboptimal treatment outcomes. Identifying genetic defects provides a unique opportunity to apply precise therapies targeting the type I IFN pathway. Unfortunately, our paper has some limitations. We will observe more patients with ACP5 and SAMHD1 mutations by genetic testing and describe the clinical characteristics of these patients.
Through bioinformatic analysis, we found that genetic alterations in ACP5 and SAMHD1 may synergistically enhance the interferon response and participate in the pathogenesis of SLE. However, this patient acquired three germline mutations from her parents. Her mother was normal but carried two mutations in ACP5 and SAMHD1, respectively. Another missense mutation in ACP5 was passed to her by her father. Since these genes are autosomal recessive, the genetic predisposition may come mainly from the double-stranded mutation in ACP5. To clarify whether the pathogenic effect is from a double-copy mutation in ACP5 or a combined defect in ACP5 and SAMHD1 resulting in an enhanced interferon response, we will perform additional functional tests to support our findings directly.
In conclusion, after a comprehensive clinical examination and high-throughput genetic testing, we diagnosed three novel missense mutations in ACP5 and SAMHD1 in a patient with refractory SLE. We analyzed the underlying pathogenesis in this patient, pointing to dysregulated IFN signaling and other indicated immune pathways. These mutations have not been previously reported and should be considered for early and accurate diagnosis of SLE. And the association of dysfunctional APC5 and SAMHD1 with immune abnormalities suggests a potential precise treatment targeting specific pathways.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Renji Hospital Biobank is funded by the National Human Genetic Resources Sharing Service Platform (2005DKA21300). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
S-MH and WC were responsible for article writing, methodology, and genetic interpretation. JF: genetic testing. DD and NS provide writing ideas and guidance. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the National Natural Science Foundation of China (31630021, 31930037, 82071843, and 81901637), Shenzhen Science and Technology Project JCYJ20180504170414637 and JCYJ20180302145033769, Shenzhen Futian Public Welfare Scientific Research Project FTWS2021006 and Sanming Project of Medicine in Shenzhen SZSM201602087, Shanghai Municipal Key Medical Center Construction Project (2017ZZ01024-002).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We gratefully acknowledge the family members for their contribution to this work. We also thank Dr. Qu Bo from Shanghai Jiao Tong University School of Medicine for his critical and insightful suggestions about our work.
References
1. Postal M, Vivaldo JF, Fernandez-Ruiz R, Paredes JL, Appenzeller S, Niewold TB. Type I interferon in the pathogenesis of systemic lupus erythematosus. Curr Opin Immunol. (2020) 67:87–94. doi: 10.1016/j.coi.2020.10.014
2. Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. (1982) 25:1271–7. doi: 10.1002/art.1780251101
3. Hochberg MC. Updating the American College of rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. (1997) 40:1725. doi: 10.1002/art.1780400928
4. Psarras A, Alase A, Antanaviciute A, Carr IM, Md Yusof MY, Wittmann M, et al. Functionally impaired plasmacytoid dendritic cells and non-haematopoietic sources of type I interferon characterize human autoimmunity. Nat Commun. (2020) 11:6149. doi: 10.1038/s41467-020-19918-z
5. Ha E, Bae SC, Kim K. Recent advances in understanding the genetic basis of systemic lupus erythematosus. Semin Immunopathol. (2022) 44:29–46. doi: 10.1007/s00281-021-00900-w
6. Jeong DY, Lee SW, Park YH, Choi JH, Kwon YW, Moon G, et al. Genetic variation and systemic lupus erythematosus: a field synopsis and systematic meta-analysis. Autoimmun Rev. (2018) 17:553–66. doi: 10.1016/j.autrev.2017.12.011
7. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. (2009) 461:747–53. doi: 10.1038/nature08494
8. Demirkaya E, Sahin S, Romano M, Zhou Q, Aksentijevich I. New horizons in the genetic etiology of systemic lupus erythematosus and lupus-like disease: monogenic lupus and beyond. J Clin Med. (2020) 9:712. doi: 10.3390/jcm9030712
9. Moser KL, Kelly JA, Lessard CJ, Harley JB. Recent insights into the genetic basis of systemic lupus erythematosus. Genes Immun. (2009) 10:373–9. doi: 10.1038/gene.2009.39
10. Eleftheriou D, Brogan PA. Genetic interferonopathies: an overview. Best Pract Res Clin Rheumatol. (2017) 31:441–59. doi: 10.1016/j.berh.2017.12.002
11. d'Angelo DM, Di Filippo P, Breda L, Chiarelli F. Type I interferonopathies in children: an overview. Front Pediatr. (2021) 9:631329. doi: 10.3389/fped.2021.631329
12. Comino EJ, Harris E, Page J, McDonald J, Harris MF. The 45 and up study: a tool for local population health and health service planning to improve integration of healthcare. Public Health Res Pract. (2016) 26:2631629. doi: 10.17061/phrp2631629
13. Banks E, Redman S, Jorm L, Armstrong B, Bauman A, Beard J, et al. Cohort profile: The 45 and up study. Int J Epidemiol. (2008) 37:941–7. doi: 10.1093/ije/dym184
14. Lacaze P, Woods R, Zoungas S, McNeil J. The genomic potential of the aspirin in reducing events in the elderly and statins in reducing events in the elderly studies. Intern Med J. (2017) 47:461–3. doi: 10.1111/imj.13384
15. Chu Y, Jiang H, Ju J, Li Y, Gong L, Wang X, et al. A metabolomic study using Hplc-Tof/Ms coupled with ingenuity pathway analysis: intervention effects of rhizoma alismatis on spontaneous hypertensive rats. J Pharm Biomed Anal. (2016) 117:446–52. doi: 10.1016/j.jpba.2015.09.026
16. Li K, He Z, Wang X, Pineda M, Chen R, Liu H, et al. Apigenin C-Glycosides of microcos paniculata protects lipopolysaccharide induced apoptosis and inflammation in acute lung injury through Tlr4 signaling pathway. Free Radic Biol Med. (2018) 124:163–75. doi: 10.1016/j.freeradbiomed.2018.06.009
17. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. String V10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. (2015) 43(Database issue):D447–52. doi: 10.1093/nar/gku1003
18. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. (2003) 13:2498–504. doi: 10.1101/gr.1239303
19. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. (2003) 4:2. doi: 10.1186/1471-2105-4-2
20. Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, et al. Cluego: a cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. (2009) 25:1091–3. doi: 10.1093/bioinformatics/btp101
21. Ravenscroft JC, Suri M, Rice GI, Szynkiewicz M, Crow YJ. Autosomal dominant inheritance of a heterozygous mutation in Samhd1 causing familial chilblain lupus. Am J Med Genet A. (2011) 155a:235–7. doi: 10.1002/ajmg.a.33778
22. Dasdemir S, Yildiz M, Celebi D, Sahin S, Aliyeva N, Haslak F, et al. Genetic screening of early-onset patients with systemic lupus erythematosus by a targeted next-generation sequencing gene panel. Lupus. (2022) 31:330–7. doi: 10.1177/09612033221076733
23. Briggs TA, Rice GI, Daly S, Urquhart J, Gornall H, Bader-Meunier B, et al. Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat Genet. (2011) 43:127–31. doi: 10.1038/ng.748
24. Fazzi E, Cattalini M, Orcesi S, Tincani A, Andreoli L, Balottin U, et al. Aicardi-goutieres syndrome, a rare neurological disease in children: a new autoimmune disorder? Autoimmun Rev. (2013) 12:506–9. doi: 10.1016/j.autrev.2012.08.012
25. Lausch E, Janecke A, Bros M, Trojandt S, Alanay Y, De Laet C, et al. Genetic deficiency of tartrate-resistant acid phosphatase associated with skeletal dysplasia, cerebral calcifications and autoimmunity. Nat Genet. (2011) 43:132–7. doi: 10.1038/ng.749
26. Navarro V, Scott C, Briggs TA, Barete S, Frances C, Lebon P, et al. Two further cases of spondyloenchondrodysplasia (Spencd) with immune dysregulation. Am J Med Genet A. (2008) 146a:2810–5. doi: 10.1002/ajmg.a.32518
27. Bilginer Y, Düzova A, Topaloglu R, Batu ED, Boduroglu K, Güçer S, et al. Three cases of spondyloenchondrodysplasia (Spencd) with systemic lupus erythematosus: a case series and review of the literature. Lupus. (2016) 25:760–5. doi: 10.1177/0961203316629000
28. Renella R, Schaefer E, LeMerrer M, Alanay Y, Kandemir N, Eich G, et al. Spondyloenchondrodysplasia with spasticity, cerebral calcifications, and immune dysregulation: clinical and radiographic delineation of a pleiotropic disorder. Am J Med Genet A. (2006) 140:541–50. doi: 10.1002/ajmg.a.31081
29. Girschick H, Wolf C, Morbach H, Hertzberg C, Lee-Kirsch MA. Severe immune dysregulation with neurological impairment and minor bone changes in a child with spondyloenchondrodysplasia due to two novel mutations in the Acp5 Gene. Pediatr Rheumatol Online J. (2015) 13:37. doi: 10.1186/s12969-015-0035-7
30. Kara B, Ekinci Z, Sahin S, Gungor M, Gunes AS, Ozturk K, et al. Monogenic lupus due to spondyloenchondrodysplasia with spastic paraparesis and intracranial calcification: case-based review. Rheumatol Int. (2020) 40:1903–10. doi: 10.1007/s00296-020-04653-x
31. Kulkarni ML, Baskar K, Kulkarni PM. A syndrome of immunodeficiency, autoimmunity, and spondylometaphyseal dysplasia. Am J Med Genet A. (2007) 143a:69–75. doi: 10.1002/ajmg.a.31526
32. Hong SW, Huh KH, Lee JK, Kang JH. Craniofacial anomalies associated with spondyloenchondrodysplasia: two case reports. Medicine (Baltimore). (2018) 97:e13644. doi: 10.1097/MD.0000000000013644
33. Sait H, Gangadharan H, Gupta A, Aggarwal A, Jain M, Phadke SR. Monogenic lupus with iga nephropathy caused by spondyloenchondrodysplasia with immune dysregulation. Indian J Pediatr. (2021) 88:819–23. doi: 10.1007/s12098-020-03636-x
34. Briggs TA, Rice GI, Adib N, Ades L, Barete S, Baskar K, et al. Spondyloenchondrodysplasia due to mutations in Acp5: a comprehensive survey. J Clin Immunol. (2016) 36:220–34. doi: 10.1007/s10875-016-0252-y
35. Zhong LQ, Wang L, Song HM, Wang W, Wei M, He YY. Spondyloenchondrodysplasia with immune dysregulation: a case report and literature review. Zhonghua Er Ke Za Zhi. (2018) 56:611–6. doi: 10.3760/cma.j.issn.0578-1310.2018.08.011
36. Dale RC, Tang SP, Heckmatt JZ, Tatnall FM. Familial systemic lupus erythematosus and congenital infection-like syndrome. Neuropediatrics. (2000) 31:155–8. doi: 10.1055/s-2000-7492
37. Aicardi J, Goutières F. Systemic lupus erythematosus or aicardi-goutières syndrome? Neuropediatrics. (2000) 31:113. doi: 10.1055/s-2000-7533
38. De Laet C, Goyens P, Christophe C, Ferster A, Mascart F, Dan B. Phenotypic overlap between infantile systemic lupus erythematosus and aicardi-goutières syndrome. Neuropediatrics. (2005) 36:399–402. doi: 10.1055/s-2005-873058
39. Rasmussen M, Skullerud K, Bakke SJ, Lebon P, Jahnsen FL. Cerebral thrombotic microangiopathy and antiphospholipid antibodies in aicardi-goutieres syndrome–report of two sisters. Neuropediatrics. (2005) 36:40–4. doi: 10.1055/s-2004-830532
40. Ramantani G, Häusler M, Niggemann P, Wessling B, Guttmann H, Mull M, et al. Aicardi-goutières syndrome and systemic lupus erythematosus (Sle) in a 12-year-old boy with Samhd1 mutations. J Child Neurol. (2011) 26:1425–8. doi: 10.1177/0883073811408310
Keywords: novel mutation, ACP5/TRAP, SAMHD1, systemic lupus erythematosus (SLE), whole-exome sequencing (WES), bioinformatics
Citation: Hong S-M, Chen W, Feng J, Dai D and Shen N (2022) Novel Mutations in ACP5 and SAMHD1 in a Patient With Pediatric Systemic Lupus Erythematosus. Front. Pediatr. 10:885006. doi: 10.3389/fped.2022.885006
Received: 27 February 2022; Accepted: 18 April 2022;
Published: 13 May 2022.
Edited by:
Mike Mikailov, United States Food and Drug Administration, United StatesReviewed by:
Mikhail Kostik, Saint Petersburg State Pediatric Medical University, RussiaSezgin Sahin, Istanbul University-Cerrahpasa, Turkey
Copyright © 2022 Hong, Chen, Feng, Dai and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nan Shen, bmFuc2hlbnNpYnNAZ21haWwuY29t; Dai Dai, ZGFpZGFpc2lic0AxNjMuY29t
†These authors have contributed equally to this work and share first authorship