
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr., 30 May 2022
Sec. Pediatric Hematology and Hematological Malignancies
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.874771
This article is part of the Research TopicInsights in Pediatric Hematology and Hematological Malignancies: 2021View all 3 articles
 Gabriel Levy1,2,3*
Gabriel Levy1,2,3* Michal Kicinski4
Michal Kicinski4 Jona Van der Straeten5Anne Uyttebroeck6Alina Ferster7
Jona Van der Straeten5Anne Uyttebroeck6Alina Ferster7 Barbara De Moerloose8Marie-Francoise Dresse9Christophe Chantrain10Bénédicte Brichard3
Barbara De Moerloose8Marie-Francoise Dresse9Christophe Chantrain10Bénédicte Brichard3 Marleen Bakkus5
Marleen Bakkus5High-throughput sequencing (HTS) of the immunoglobulin heavy chain (IgH) locus is a recent very efficient technique to monitor minimal residual disease of B-cell precursor acute lymphoblastic leukemia (BCP-ALL). It also reveals the sequences of clonal rearrangements, therefore, the multiclonal structure, of BCP-ALL. In this study, we performed IgH HTS on the diagnostic bone marrow of 105 children treated between 2004 and 2008 in Belgium for BCP-ALL in the European Organization for Research and Treatment of Cancer (EORTC)-58951 clinical trial. Patients were included irrespectively of their outcome. We described the patterns of clonal complexity at diagnosis and investigated its association with patients’ characteristics. Two indicators of clonal complexity were used, namely, the number of foster clones, described as clones with similar D-N2-J rearrangements but other V-rearrangement and N1-joining, and the maximum across all foster clones of the number of evolved clones from one foster clone. The maximum number of evolved clones was significantly higher in patients with t(12;21)/ETV6:RUNX1. A lower number of foster clones was associated with a higher risk group after prephase and t(12;21)/ETV6:RUNX1 genetic type. This study observes that clonal complexity as accessed by IgH HTS is linked to prognostic factors in childhood BCP-ALL, suggesting that it may be a useful diagnostic tool for BCP-ALL status and prognosis.
1. IgH high-throughput sequencing allows new insights into the clonal architecture of BCP-ALL.
2. A higher number of evolved clones at diagnosis of BCP-ALL was associated with the presence of t(12;21)/ETV6:RUNX1.
3. Patients with a higher number of foster clones were patients in the better prognosis group.
B-cell precursor acute lymphoblastic leukemia (BCP-ALL) is the most common pediatric neoplasm (1, 2). It is a clonal genetic heterogeneous disease generally thought to arise from the malignant transformation and expansion of a single lymphoid progenitor at various stages of development (3–5). The precise pathogenetic events leading to the development of ALL are still unknown, but evidence supports the hypothesis of driver mutations followed by secondary events, that can occur in subclones of the original leukemic cell following different evolution patterns (5, 6).
Early in B-cell development, somatic recombinations at the immunoglobulin heavy chain (IgH) locus give rise to unique rearrangements resulting from the random coupling between one of the many possible variable (VH), diversity (D), and joining (JH) genes [V(D)J recombination or combinatorial diversity], as well as imprecise joining of gene segments and the addition of nucleotides to the DNA sequence at splice sites (N-diversity or junctional diversity) (Figure 1) (7, 8). Identical IgH rearrangements, which are a unique signature to B-cells, reflect the clonal nature of a population, and reversely, the clonality of B-cell populations can be assessed by IgH analysis. By extension, IgH rearrangements constitute clonotypic markers and allow high-resolution tracking of the architecture and clonal dynamic of BCP-ALL cells (3, 9).
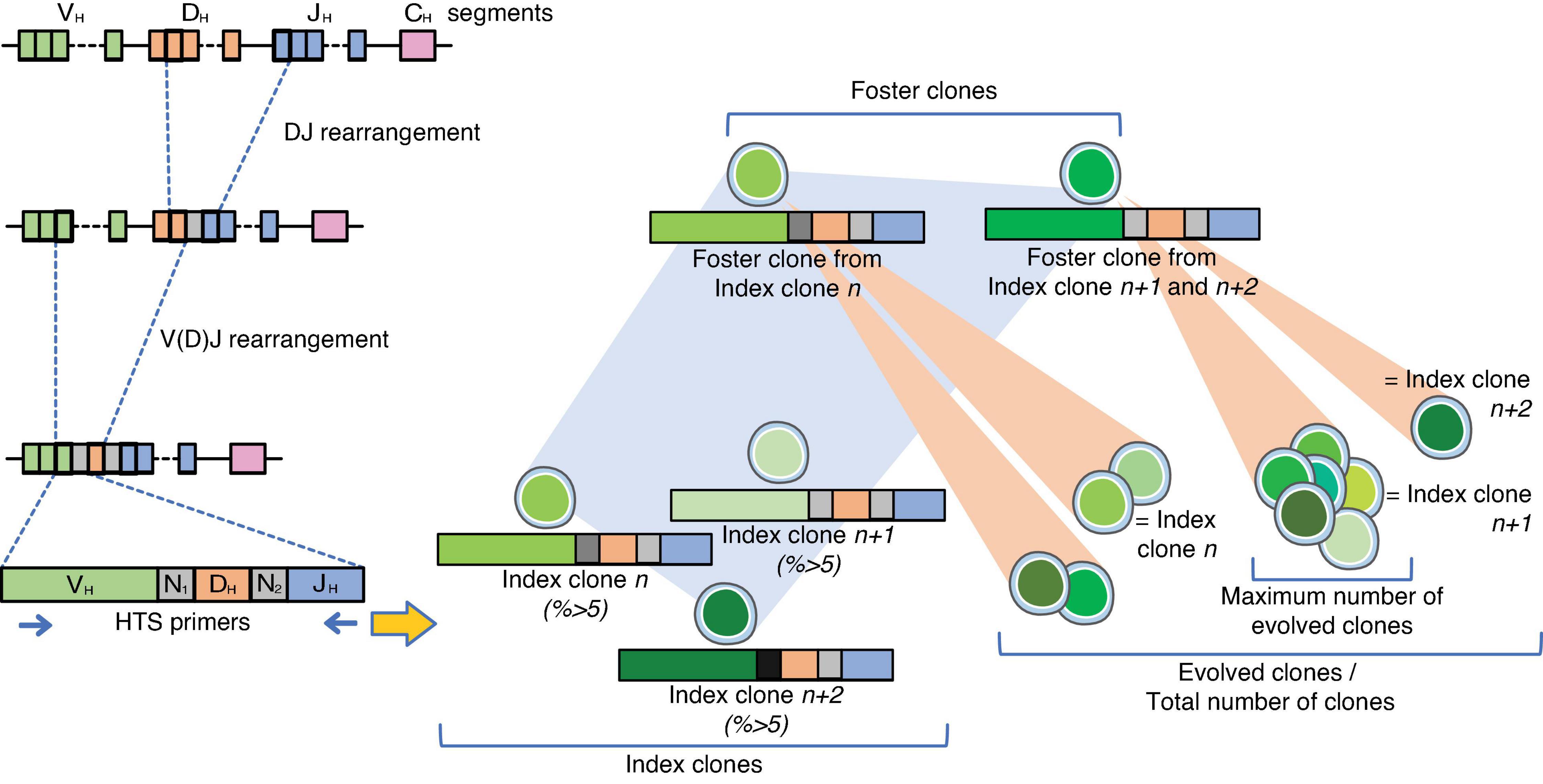
Figure 1. Design of the experiments. On the left panel, the steps for the V(D)J somatic recombination of the IgH locus are represented. Diversity is enabled through a random combination of one of many variables (V), diversity (D), and joining (J) gene segments although the major contributor to the diversity of the immunoglobulin repertoire is the variable truncation of the recombined gene segments in synergy with the addition of non-templated (N) nucleotides, the so-called N regions within the rearrangements (8). Index clones were designated as clones representing ≥5% of the individual clonotypes with the same V(D)J rearrangement. Analysis of the V(D)J-sequence allowed identification of related foster clones among index clones. Foster clones were defined as clones with similar D-N2-J rearrangements, but other V-rearrangements and N1-joining, regardless of their percentage. Evolved clones were clones related to the index and foster clones by sharing the same or partly the same D-J stem, regardless of their frequency. The total number of clones per patient was the sum of evolved clones, and the maximum number of evolved clones was the highest number of evolved clones across all foster clones.
In fact, IgH studies in paired diagnosis, treatment follow-up, and relapse samples revealed that leukemic cells maintain ongoing IgH changes alongside the disease (10, 11), in particular VH replacement. These changes give insight into the continuous evolution of the BCP-ALL structure with a given number of leukemic subclones that are present at diagnosis or can appear during treatment and possibly reemerge at relapse alongside a dominant clone (3, 9, 10).
Minimal residual disease (MRD), reflecting treatment efficiency, is considered to be the strongest prognostic factor in both children and adult ALL, independently of traditional prognostic factors, such as age, blast count at diagnosis, immunophenotype, or genetic abnormalities (12–15). The term MRD describes a level of disease that is undetectable by conventional cytomorphology and is not accompanied by any clinical symptom. Current methods to monitor MRD in ALL include multicolor flow cytometric (MFC) detection of aberrant immunophenotypes, allele-specific oligonucleotide RQ-PCR (ASO-PCR) amplification of immunoglobulin (Ig), and T-cell receptor (TCR) genes and/or real-time quantitative polymerase chain reaction (RQ-PCR) of fusion transcripts (16–19). Although MFC, ASO-, or RQ-PCR methods are used in a clinical setting and have proven reliable to reach high sensitivity, they all have their own limitations. Among those, MFC can lead to a false-negative finding if antigen expression changes over the course of the disease, fusion transcript RQ-PCR is applicable only in patients with target fusion genes and faces limited standardization, and Ig/TCR ASO-PCR is time-consuming as it needs optimization of patient-specific reagents and assays, which themselves are prone to false-negative results following clonal evolution of the disease (16, 18).
Next-generation sequencing (NGS)-based methods, also called deep- or high-throughput sequencing (HTS) methods, can be used to monitor MRD by detailed sequencing of the V(D)J junctions (16, 17, 20–23).
Next-generation sequencing methods have the advantage to allow quick access to the full IgH repertoire of an individual, without the necessity to develop personalized assays, and have proven to give a more complete insight into the leukemic population than conventional ASO-PCR at each time-point of the MRD monitoring. In comparison to current methods of MRD measurement that had limited to no capacity to monitor the evolution of leukemic subclones during treatment, they thus allow follow-up of subclones and/or identification of new emerging clones throughout the evolution of the disease (3, 21, 24).
Therefore, these methods are sources of great promises, and their major limitation in children resides currently in the need for standardized bioinformatics methods to interpret thoroughly the results of cohort studies to validate this approach (16, 23).
Along with their development for monitoring MRD, HTS methods have also shed light on mechanisms associated with leukemic clonal evolution that were previously underappreciated and have allowed studying leukemic cell evolution according to their niche. For example, Bartram et al. were able to demonstrate that central nervous system (CNS) and bone marrow (BM) clones are the same alongside the evolution of the disease and that BM infiltration would be present at some level, even in apparently isolated CNS relapse (25).
Comprehension of the molecular pathways involved in V(D)J recombinations is still being investigated, as is their relation with genetic instability and potential oncogenicity (26). Papaemmanuil et al. demonstrated in 2014 the relation between leukemogenesis in t(12;21)/ETV6:RUNX1 positive childhood BCP-ALL leukemia and the RAG recombinase activity, an endonuclease required for V(D)J recombination (27). Furthermore, RAG-mediated aberrant recombinations might also be involved in the evolution of t(9;22)/BCR-ABL positive BCP-ALL (28, 29).
Nevertheless, to date, there is less understanding of the association between the clonal architecture of the disease at diagnosis and its clinical presentation or prognosis. In this study, we investigated the association between the number of leukemic clones, as defined by clonal IgH rearrangements, the number of evolved clones, as defined by the same D-J stem, and the characteristics of patients and known prognostic factors of BCP-ALL at diagnosis.
The samples of this study originated from patients registered in the European Organization for Research and Treatment of Cancer (EORTC)-58951 study for the treatment of ALL or lymphoblastic non-Hodgkin’s lymphoma in children between one and 18 years (Supplementary Figure 1) (30, 31).
A subgroup of Belgian patients treated for BCP-ALL between 2004 and 2008 was systematically reviewed for IgH recombinations. Diagnostic BM samples with a clonal IgH rearrangement and sufficient leftover material were retained for the study. The patients were selected with no knowledge of their outcomes. A total of 105 samples that fulfilled inclusion criteria were used for NGS analysis.
Mononuclear cells (MNCs) had been separated by Lymphoprep™ density gradient centrifugation (Elitech – Cat. No. AX-1114547) from BM aspirates at diagnosis, and genomic DNA was extracted using the QIAamp® DNA Blood Mini Kit (QIAGEN®). The DNA was stored at −20°C for later use.
Next-generation sequencing for IgH was performed using the LymphoTrack® IGH FR1-MiSeq® kit (Invivoscribe®, Cat. No. 91210039). A maximum of 50 ng diagnostic DNA was amplified and sequenced. The primers used for sequencing targeted the framework region 1 (FR1) and the JH region. The library was sequenced with the MiSeq® device (Illumina®, 2 × 250 cycles) at a final concentration of 14 pM and 1% PhiX. A minimum of 20.000 reads had to be obtained for each sample. Sequencing results were aligned with IMGT/V-QUEST (7).
Index clones: the frequency of every clonotype in each sample was determined by calculating the number of sequencing reads for each clonotype divided by the total number of sequencing reads in the sample. In line with former studies (3, 32), an index clone was designated as a clone representing ≥5% of the individual clonotypes (Figure 1).
Foster clones: analysis of the V(D)J-sequence allowed identification of related foster clones among index clones. Foster clones were defined as clones with similar D-N2-J rearrangements, but other V-rearrangement and N1-joining, regardless of their percentage. The foster clones were manually sorted by searching all the sequences with the same D-N2-J region; a minimum of 1/2 of the D-region was used. In case of no D-region, a minimum of 1/2 of the N-region was used. A foster clone indicator was given to each foster clone.
Evolved clones: clones related to the foster clone by sharing the same or partly the same D-J stem, regardless of their frequency. The total number of evolved clones per patient was defined as the sum of clones across all index clones, where the number of clones per index clone was equal to the number of clones evolved from the index clone +1 (as the number of evolved clones did not include the original index clone).
To abrogate the arbitrary 5%-limit designating the index clones, two indicators of clonal complexity were used for the statistical analysis:
(1) The number of foster clones.
(2) The maximum across all foster clones of the number of evolved clones from one foster clone (referred to as the maximum number of evolved clones hereafter). This number included the foster clone itself.
The following covariates were considered in the description of the population and for the assessment of prognostic categorization: sex, age at diagnosis (1 to <5 vs. 5 to <10 vs. ≥ 10 years), white blood cell (WBC) count at diagnosis (<10 × 109/L vs. 10 × 109 to <50 × 109/L vs. ≥50 × 109/L), initial CNS involvement (CNS-1 vs. others) (30, 31), National Cancer Institute (NCI) risk group (33), EORTC risk group after prephase, and ALL genetic type. EORTC risk group was defined as in the 58951 trial analysis [initial very low risk (VLR) vs. average risk 1 (AR1) vs. AR2 vs. very high risk (VHR)] (30, 31). ALL genetic type was classified as: t(12;21)/ETV6:RUNX1 abnormality determined by FISH vs. hyperdiploidy vs. others. Hyperdiploidy (yes vs. no) was defined as in previous EORTC studies (>50 chromosomes, DNA index, and FISH used in case of no reliable cytogenetic data available) (34).
We studied the association between clonal complexity as indicated by the number of foster clones and the maximum number of evolved clones and other patients’ characteristics. For the continuous covariates (age and WBC count) and the ordinal covariate EORTC risk group (VLR vs. AR1 vs. AR2 vs. VHR), the Spearman test was used. For the covariates with two categories (sex, NCI risk group, and initial CNS involvement defined as CNS-1 vs. other) and the nominal covariate ALL genetic type [t(12;21)/ETV6:RUNX1 vs. hyperdiploidy vs. other], the Kruskal–Wallis test was used. All tests were performed at a two-sided significance level of 0.05. The analysis was performed using SAS version 9.4.
Agreement for clinical and biological research according to local and international guidelines had been issued at the time of inclusion in the EORTC-58951 clinical trial. DNA samples from leukemic cells were issued from archived material issued for diagnostic and follow-up purposes.
The description of the population is summarized in Table 1. Among the patients from the EORTC-58951 study, 105 who entered the trial between 2004 and 2008 meeting the inclusion criteria of BCP-ALL with the target for the IgH study were selected (Supplementary Figure 1). There were 75 patients (71.4%) with NCI standard-risk leukemia and 30 (28.6%) with NCI high-risk leukemia. According to the EORTC risk group, 23 patients (21.9%) were in the VLR group, 63 (60.0%) in the AR1 group, 9 (8.6%) in the AR2 group, and 10 (9.5%) in the VHR group. Leukemia involved t(12;21)/ETV6:RUNX1 abnormality in 27 cases (25.7%) and hyperdiploidy in 39 cases (38.6%).

Table 1. Description of the population.
The number of index clones was 1 for 24.8%, 2 for 45.7%, 3 for 23.8%, and greater than 3 for 5.7% of the patients (Table 2 and Supplementary Table 1). The number of foster clones was 1 for 27.6%, 2 for 55.2%, 3 for 16.2%, and 4 for 1.0% of the patients. The median of the total number of evolved clones per patient was 13 (range: 1–1,046). The median number of the maximum number of evolved clones was 9 (range: 1–734). The maximum number of evolved clones was weakly associated with the number of foster clones (Spearman correlation: rho = 0.20, p = 0.04) (Figure 2A). On the log scale, there was a strong linear association between the total number of clones and the maximum number of evolved clones, indicating that among patients with many evolved clones, the clones typically evolved from the same foster clone (Figure 2B).
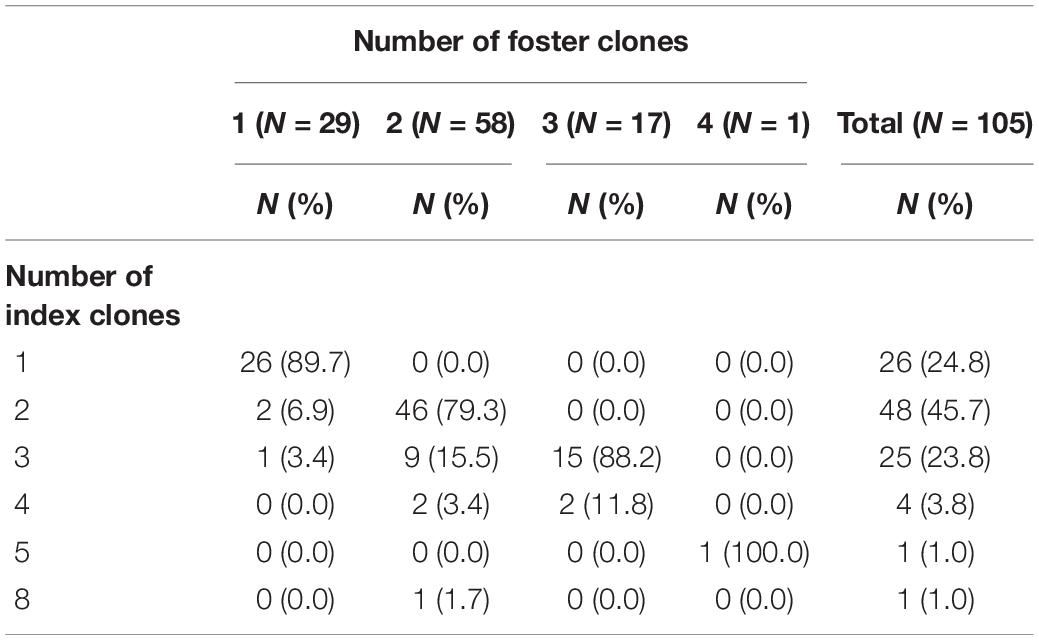
Table 2. Distribution of the number of index clones and foster clones.
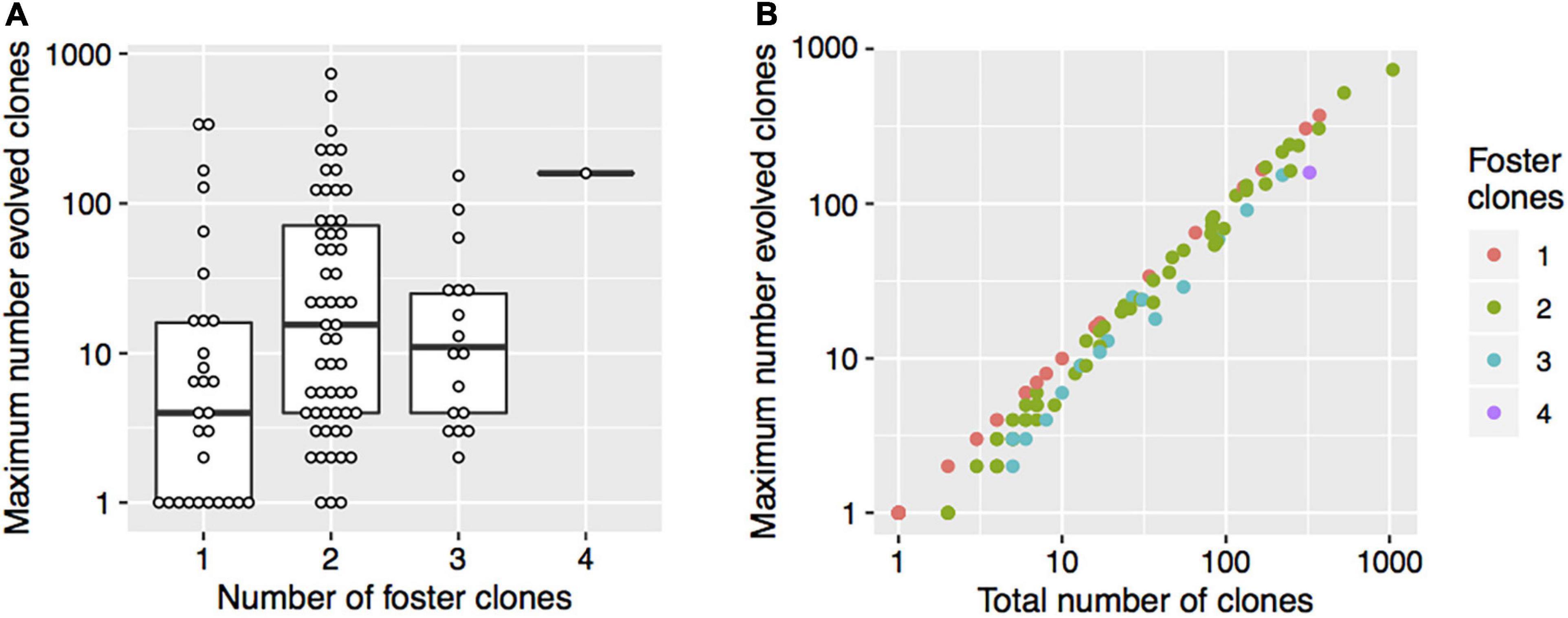
Figure 2. The number of foster clones, the maximum number of evolved clones, and the total number of clones. (A) The thick horizontal lines represent the medians and the boxes indicate the first and the third quartiles of the maximum number of evolved clones. Each point shows data for one patient. (B) The colors indicate the number of foster clones. Each point shows data for one patient.
We found no significant association between the number of foster clones and the covariates sex, age, WBC count at diagnosis, initial CNS involvement, or NCI risk group (Table 3). Patients in lower EORTC risk groups had more foster clones than patients in higher-risk groups (p = 0.007). The genetic type was associated with the number of foster clones as well (p = 0.032), with patients with t(12;21)/ETV6:RUNX1 having a smaller number of foster clones. Among patients with t(12;21)/ETV6:RUNX1, 7.4% of the patients had three or four clones, as compared with 25.6% among patients with hyperdiploidy and 17.1% of patients with other genetic types.
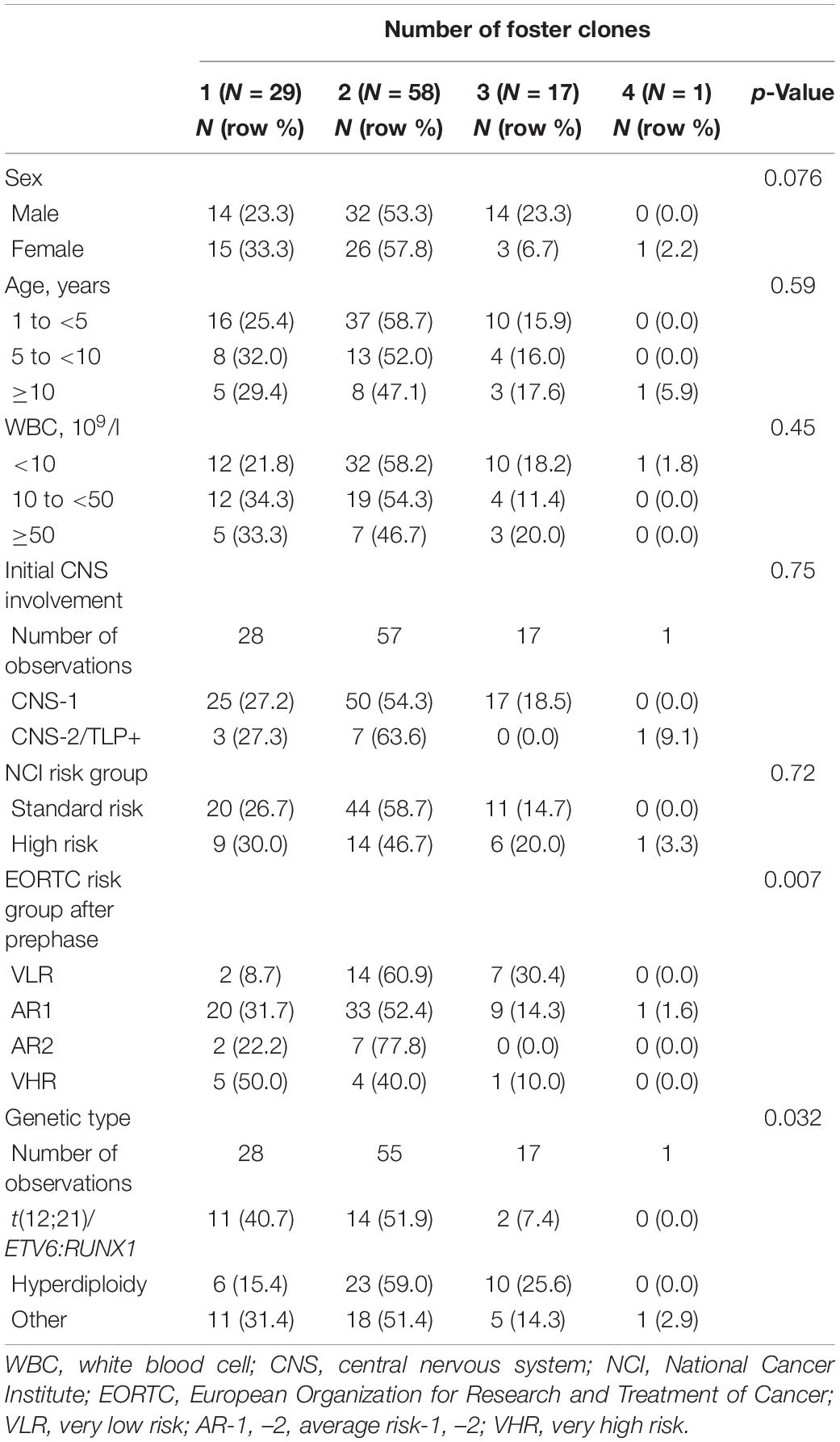
Table 3. Associations between the number of foster clones and patients’ characteristics.
There was no significant association between the covariates sex, age, WBC count at diagnosis, NCI, EORTC risk group, or initial CNS involvement and the maximum number of evolved clones (Table 4). However, the maximum number of evolved clones was strongly associated with the genetic type (p = 0.002) (Table 4 and Figure 3). Patients with t(12;21)/ETV6:RUNX1 had significantly more evolved clones than patients with hyperdiploidy or other genetic abnormalities. The median of the maximum number of evolved clones was 54 for t(12;21)/ETV6:RUNX1, 5 for hyperdiploidy, and 6 for other genetic types.
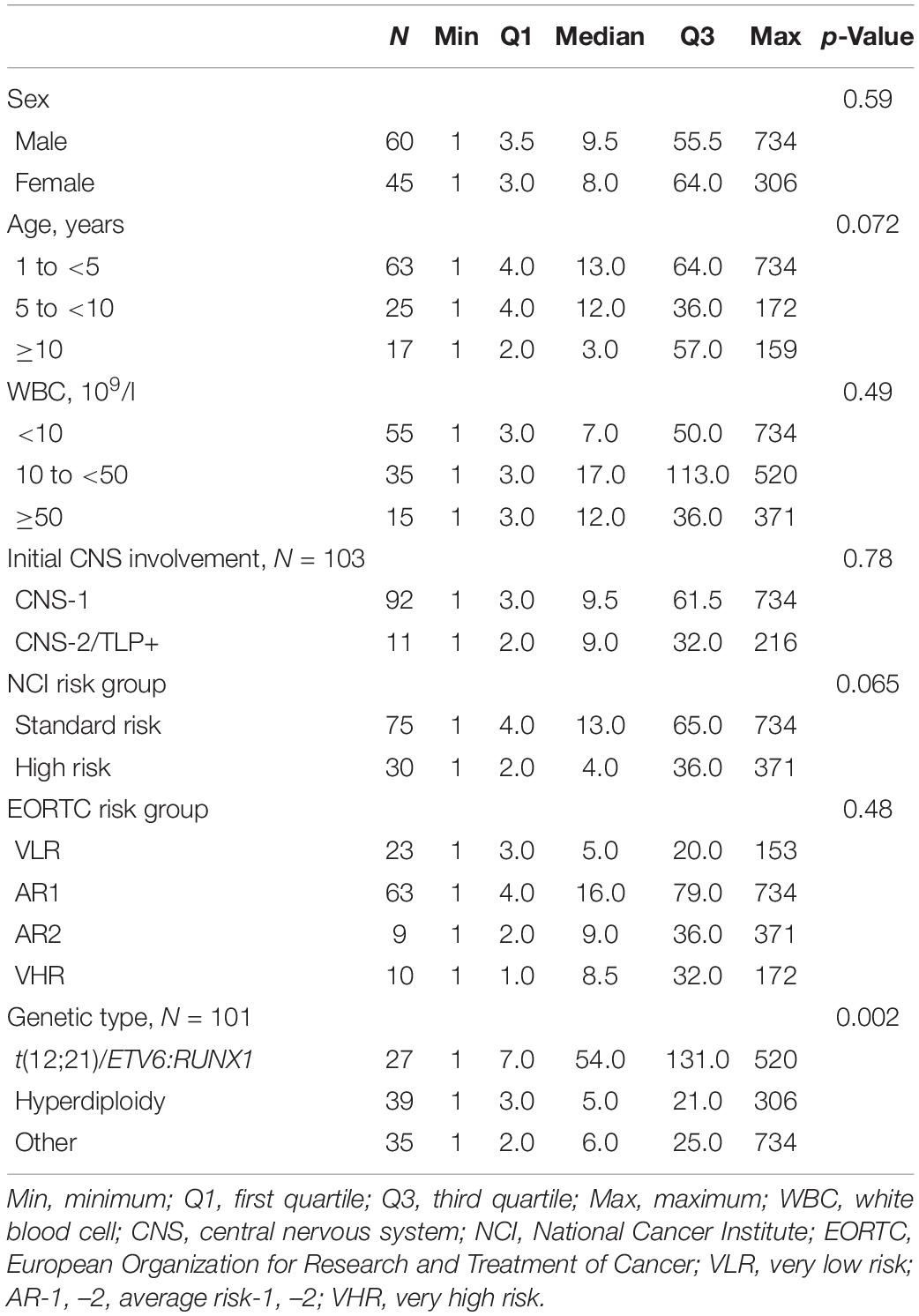
Table 4. Associations between the maximum number of evolved clones and patients’ characteristics.
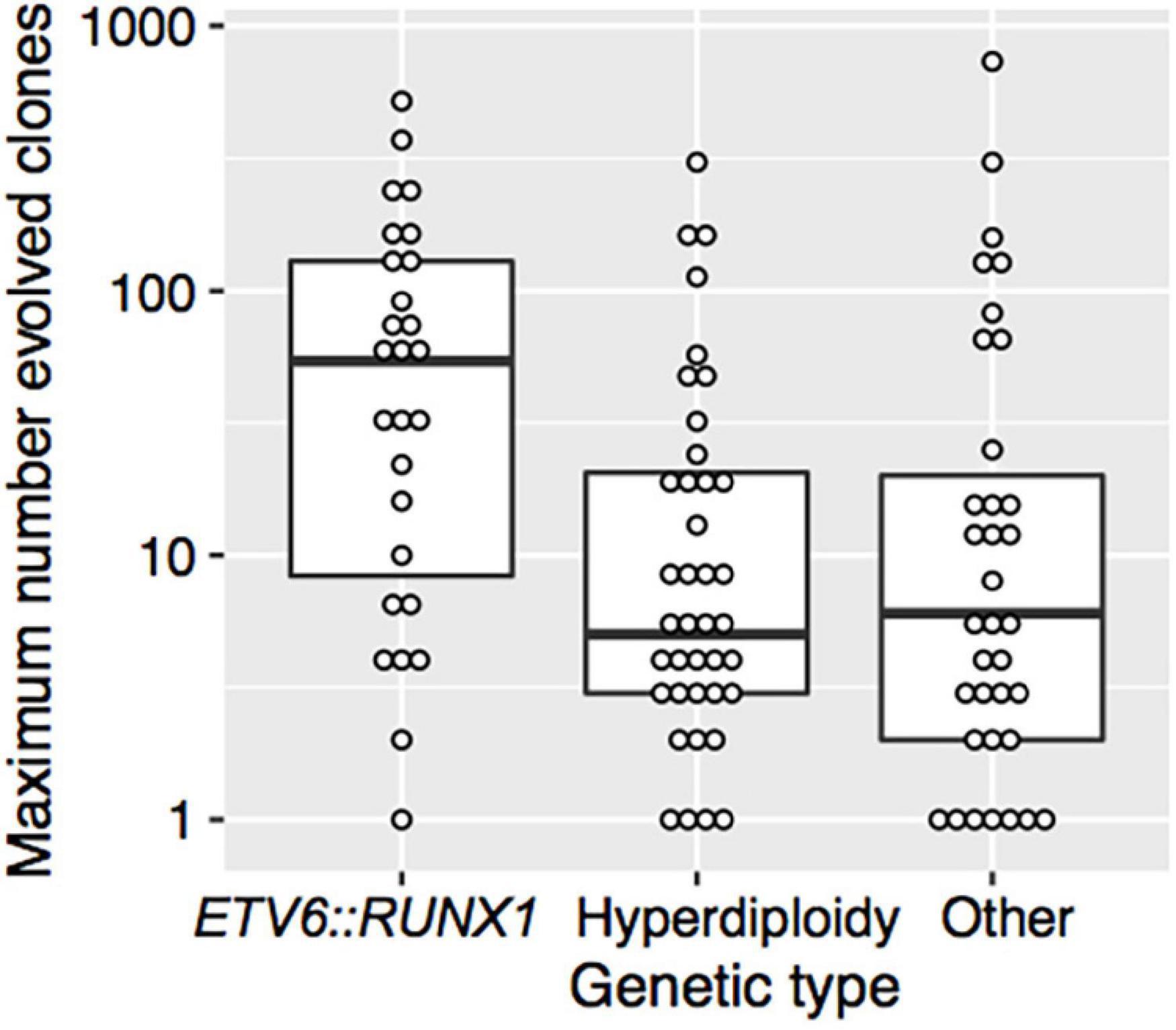
Figure 3. The maximum number of evolved clones vs. genetic type. The thick horizontal lines represent the medians and the boxes indicate the first and the third quartiles of the maximum number of evolved clones. Each point shows data for one patient.
To analyze the correlation between clonal complexity of leukemia at diagnosis and described prognostic risk factors, we chose in this study the categories of foster clones and evolved clones from the foster clone. The notion of index clone, as described since the first studies on IgH HTS, takes indeed separately into account each clone with a frequency higher than 5% (3). By manually analyzing the D-N2-J sequences, we could gather index clones belonging together, sharing the same D-N2-J stem, thus originating from the same foster clone. Then, manual sorting allowed us to describe precisely the number of evolved clones, regardless of their percentage. The numbers of foster clones and of evolved clones from the foster clones were significantly associated. Leukemia with a higher number of foster clones had also more evolved clones.
A majority of patients had two foster clones. This observation was enabled by the fact that we considered the foster clones rather than the index clones and would be in line with Alves-Pereira et al. (35), who showed in 2014 that both IgH alleles are recruited independently and in parallel during V(D)J recombination in pre-B cells. This process of rearrangement is regulated by feedback mechanisms that are set up once a productive VH to DJH joining took place and which are partly lacking in leukemic cells (11). It has also been suggested that the pattern of ongoing rearrangements in an individual patient reflects the IgH rearrangement status of the precursor cell at the time of malignant transformation (11). As the idea of a monoclonal origin of ALL is nowadays undermined by many studies that reported that up to 40% of BCP-ALL are (at least) oligoclonal at diagnosis (3, 36), this might explain why 18 patients had more than two foster clones (17%). Furthermore, it is not known what proportion of cells rearrange the alleles synchronously (35), which could also account for leukemic cell lines with only one foster clone.
In patients with t(12;21)/ETV6:RUNX1, there were significantly less foster clones but the maximum number of evolved clones was higher. The t(12;21)/ETV6:RUNX1 translocation is present in around 25% of childhood BCP-ALL (5) and is related to a better prognosis (37). This would therefore be in line with the fact that patients who had more clonal evolutions also belonged more often to the EORTC VLR or AR1 risk.
That patients with t(12;21)/ETV6:RUNX1 had significantly less foster clones, and a higher maximum number of evolved clones could confirm results obtained in 2004 by Hübner et al. on t(12;21)/ETV6:RUNX1 BCP-ALL, who found that t(12;21)/ETV6:RUNX1 BCP-ALL had a higher number of Ig/TCR rearrangements but with lower IgH oligoclonality (38).
Biologically, t(12;21)/ETV6:RUNX1 would appear early in leukemic blasts (39) and lead to an arrest in B-cell differentiation but would not be sufficient to induce leukemia (29). The critical secondary events leading to leukemic transformation in t(12;21)/ETV6:RUNX1 BCP-ALL would frequently be linked to genomic rearrangements mediated by aberrant RAG recombinase activity (27), which is increased in t(12;21)/ETV6:RUNX1 BCP-ALL (40, 41). The fact that RAG activity plays an important role in V(D)J rearrangement (26, 42) might furthermore explain why we found significantly more clonal evolution in the patients who had t(12,21)/ETV6:RUNX1 positive BCP-ALL.
The RAG activity is however not only increased in t(12;21)/ETV6:RUNX1 BCP-ALL and is found, for example, in BCR-ABL1 ALL (28), which were considered of bad prognosis before the availability of tyrosine kinase inhibitors (43). ALL with KMT2A translocation in infants are also known to be oligoclonal and are of worse prognosis (36, 44). Moreover, if RAG activity would be responsible for secondary translocations in t(12;21)/ETV6:RUNX1 BCP-ALL, they seem neither to be the cause of the latter translocation nor explain early translocations in fetal life (45). Besides, the relation between the number of clones and the molecular characteristics of the leukemic cells has not been established so far and relies on much more complex and multifactorial mechanisms than RAG activity, which intervenes at the cleavage phase. In 2014, Gawad et al. (46) individually sequenced 1,479 single tumor cells from six patients with BCP-ALL. In addition to the clonal structure of the disease, they showed how deletions, IgH sequences, and specific mutations segregated between clones. They also confirmed (19, 47) that ongoing V(D)J recombination of variable magnitude between different clones in the same patient could occur in the most evolved clones. Generalization of their results might however be difficult, as five out of the six patients in the study harbored t(12;21)/ETV6:RUNX1.
In their first HTS study (3), Gawad et al. showed the multiplicity of the potential evolution of leukemic cells. We found between 1 and 1,046 evolved clones per patient in our study. This number differed greatly in different studies, between 1 and 4,025 in the study by Gawad et al. (3), between 1 and 6,934 in the study by Faham et al. (17), or between 9 and 59 in the study by Bashford-Rogers et al. (9). Nevertheless, most studies do not refer to the total number of evolved clones, and the phylogeny of the leukemic cells is not always taken into account in HTS studies. The evolution of the IgH repertoire defining the evolved clones as seen by HTS needs however to be studied deeper, as it seems to progress separately from the mutational evolution of subclones (6, 48), or even from the changing immunophenotype of subclones (46) alongside the disease and at relapse (49).
With the generalization of HTS as a new way to efficiently monitor MRD (18), there is all the more a need for the consensual definition for clones and subclones, as MRD is another independent prognostic marker of BCP-ALL, if not the most important (12, 13, 15). A European network, the EuroClonality-NGS Consortium was created to tackle these questions (23) as software are being designed and tested (20, 22, 50, 51) for monitoring MRD by HTS. This study gives insight into the problems that have to be considered whenever leukemic clones are defined by their clonal IgH sequence. The question remains whether evolved clones are part of the leukemic clone and thus have to be monitored in the MRD testing or just an epiphenomenon reflecting the maturation phase of the leukemic clone. Moreover, we based our study on index clones (and consecutively, on foster clones) defined as clones above the threshold of five percent of the individual clonotypes (3, 32). With evolving HTS technics and deeper sequencing, this definition could also vary and lead to another comprehension of the clonal landscape of leukemias.
Finally, we found no statistically significant association between the other characteristics and prognostic factors for leukemia in children (sex, age, WBC count at diagnosis, NCI risk group, or initial CNS involvement) and the number of foster clones and evolved clones. One possible explanation for this is the relatively small number of cases investigated. Although this study constitutes one of the biggest cohorts on IgH HTS at diagnosis, the sample size would not have allowed enough statistical power to analyze the association between clonal complexity and EFS or other described prognostic factors of pediatric leukemia, which is indeed a very heterogeneous disease with many prognostic factors present only in small subsets of patients, as, for example, genetic abnormalities (52, 53). Furthermore, only patients with IgH recombinations were included in this study and we did not investigate the association between the absence of IgH recombination (which stipulated an exclusion from our cohort) and belonging to a particular group of risk. Ding et al., among others, suggested that it might be interesting to also look at recombinations of the TCR, even in patients with BCP-ALL, as around 10% of their cohort of patients with BCP-ALL expressed a dominant TCR rearrangement (24 cases out of 219 patients with ALL) (49), while the so-called illegitimate rearrangements – TR rearrangements in BCP-ALL – have been identified in up to 80–90% of patients with BCP-ALL (54, 55). Likewise, some 40% of BCP-ALL also carry an IGK rearrangement (54). We focused our study on diagnostic blasts as sequencing of relapse samples was not available to us. Studies targeting the evaluation of HTS for MRD monitoring will hopefully allow gaining access to such data (20).
Furthermore, we limited our study to IgH sequencing and did not look at BCR expression. It has been shown that many ALL carry non-productive BCR/TCR (56) in both alleles or the only expressed dominant allele, which was suggested to support the hypothesis that BCR might act as a tumor suppressor in most cases of B-precursor ALL (57). This might therefore also be looked at when considering prognostic factors and IgH rearrangements.
One interesting point in our study remains that we considered the number of clones and evolved clones as a potential individual and isolated prognostic marker of the disease, a question not referred to in studies on clonality in BCP-ALL. There is to date less knowledge of the link between genetic alterations in BCP-ALL and recombinations of the IgH or TCR, although some authors suggested a role of some genetic aberrations or age at diagnosis (38, 55, 58, 59). New methods to dig into clonality as single-cell DNA amplicon sequencing (60) could help understand those mechanisms and be combined with sequencing of the IgH or TCR.
Our study does not allow the proclamation of the number of foster clones or of evolved clones from the foster clone as new and prognostic factors for childhood BCP-ALL. Further studies on a bigger scale would be needed to support this hypothesis and might end up in subclonal analyses being part of compound prognostic scores. The generalization of HTS methods for the measurement of MRD might bring opportunities to gain access to such HTS data of diagnostic and follow-up ALL samples.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://github.com/GabrielLevyUCLicr/LevyBakkusEtAl_frontiers.
The studies involving human participants were reviewed and approved by the clinical trial registered: https://clinicaltrials.gov/ct2/show/NCT00003728. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
GL, MK, BB, and MB designed the study, interpreted the data, and wrote the manuscript. JV and MB performed the experiments. MK performed the statistical analysis. AU, AF, BD, M-FD, and CC provided data on patients. All authors contributed to the article and approved the submitted version.
This study was supported by a donation from the La Fondation contre le Cancer from Belgium and from Kom op tegen Kanker (Stand Up to Cancer) and the Flemish cancer society from Belgium. MB was supported by the Kinderkankerfonds (Belgium) and Télévie grant 28597737 (Belgium).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank all the EORTC CLG members, the patients, and the clinicians who participated in the EORTC-58951 clinical trial.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.874771/full#supplementary-material
1. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med. (2015) 373:1541–52. doi: 10.1056/NEJMra1400972
2. Howlader N, Noone AM, Krapcho M, Miller D, Brest A, Yu M, et al. SEER Cancer Statistics Review, 1975-2018. (2021). Available online at: https://seer.cancer.gov/csr/1975_2018/index.html (accessed November 16, 2021).
3. Gawad C, Pepin F, Carlton VEH, Klinger M, Logan AC, Miklos DB, et al. Massive evolution of the immunoglobulin heavy chain locus in children with B precursor acute lymphoblastic leukemia. Blood. (2012) 120:4407–17. doi: 10.1182/blood-2012-05-429811
4. Pui C-H, Campana D, Evans WE. Childhood acute lymphoblastic leukaemia – current status and future perspectives. Lancet Oncol. (2001) 2:597–607. doi: 10.1016/S1470-2045(01)00516-2
5. Pui C-H, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. (2004) 350:1535–48. doi: 10.1056/NEJMra023001
6. Lindqvist CM, Lundmark A, Nordlund J, Freyhult E, Ekman D, Carlsson Almlöf J, et al. Deep targeted sequencing in pediatric acute lymphoblastic leukemia unveils distinct mutational patterns between genetic subtypes and novel relapse-associated genes. Oncotarget. (2016) 7:64071–88. doi: 10.18632/oncotarget.11773
7. Lefranc M-P, Giudicelli V, Ginestoux C, Jabado-Michaloud J, Folch G, Bellahcene F, et al. IMGT(R), the international ImMunoGeneTics information system(R). Nucleic Acids Res. (2009) 37:D1006–12. doi: 10.1093/nar/gkn838
8. Cabaniols JP, Fazilleau N, Casrouge A, Kourilsky P, Kanellopoulos JM. Most alpha/beta T cell receptor diversity is due to terminal deoxynucleotidyl transferase. J Exp Med. (2001) 194:1385–90. doi: 10.1084/jem.194.9.1385
9. Bashford-Rogers RJM, Nicolaou KA, Bartram J, Goulden NJ, Loizou L, Koumas L, et al. Eye on the B-ALL: B-cell receptor repertoires reveal persistence of numerous B-lymphoblastic leukemia subclones from diagnosis to relapse. Leukemia. (2016) 30:2312–21. doi: 10.1038/leu.2016.142
10. Szczepañski T, Willemse MJ, Brinkhof B, van Wering ER, van der Burg M, van Dongen JJ. Comparative analysis of Ig and TCR gene rearrangements at diagnosis and at relapse of childhood precursor-B–ALL provides improved strategies for selection of stable PCR targets for monitoring of minimal residual disease. Blood. (2002) 99:2315–23. doi: 10.1182/blood.V99.7.2315
11. Steenbergen EJ, Verhagen OJ, van Leeuwen EF, von dem Borne AE, van der Schoot CE. Distinct ongoing Ig heavy chain rearrangement processes in childhood B-precursor acute lymphoblastic leukemia. Blood. (1993) 82:581–9.
12. van Dongen JJ, Seriu T, Panzer-Grümayer ER, Biondi A, Pongers-Willemse MJ, Corral L, et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet. (1998) 352:1731–8. doi: 10.1016/S0140-6736(98)04058-6
13. Borowitz MJ, Devidas M, Hunger SP, Bowman WP, Carroll AJ, Carroll WL, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a children’s oncology group study. Blood. (2008) 111:5477–85. doi: 10.1182/blood-2008-01-132837
14. Borowitz MJ, Wood BL, Devidas M, Loh ML, Raetz EA, Salzer WL, et al. Prognostic significance of minimal residual disease in high risk B-ALL: a report from children’s oncology group study AALL0232. Blood. (2015) 126:964–71. doi: 10.1182/blood-2015-03-633685
15. Cavé H, van der Werff ten Bosch J, Suciu S, Guidal C, Waterkeyn C, Otten J, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia. European organization for research and treatment of cancer–childhood leukemia cooperative group. N Engl J Med. (1998) 339:591–8. doi: 10.1056/NEJM199808273390904
16. van Dongen JJ, van der Velden VH, Brüggemann M, Orfao A. Minimal residual disease diagnostics in acute lymphoblastic leukemia: need for sensitive, fast, and standardized technologies. Blood. (2015) 125:3996–4009. doi: 10.1182/blood-2015-03-580027
17. Faham M, Zheng J, Moorhead M, Carlton VEH, Stow P, Coustan-Smith E, et al. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood. (2012) 120:5173–80. doi: 10.1182/blood-2012-07-444042
18. Kotrova M, Trka J, Kneba M, Brüggemann M. Is next-generation sequencing the way to go for residual disease monitoring in acute lymphoblastic leukemia? Mol Diagn Ther. (2017) 21:481–92. doi: 10.1007/s40291-017-0277-9
19. Germano G, del Giudice L, Palatron S, Giarin E, Cazzaniga G, Biondi A, et al. Clonality profile in relapsed precursor-B-ALL children by GeneScan and sequencing analyses. Consequences on minimal residual disease monitoring. Leukemia. (2003) 17:1573–82. doi: 10.1038/sj.leu.2403008
20. Kotrova M, Muzikova K, Mejstrikova E, Novakova M, Bakardjieva-Mihaylova V, Fiser K, et al. The predictive strength of next-generation sequencing MRD detection for relapse compared with current methods in childhood ALL. Blood. (2015) 126:1045–7. doi: 10.1182/blood-2015-07-655159
21. Ferret Y, Caillault A, Sebda S, Duez M, Grardel N, Duployez N, et al. Multi-loci diagnosis of acute lymphoblastic leukaemia with high-throughput sequencing and bioinformatics analysis. Br J Haematol. (2016) 173:413–20. doi: 10.1111/bjh.13981
22. Pulsipher MA, Carlson C, Langholz B, Wall DA, Schultz KR, Bunin N, et al. IgH-V(D)J NGS-MRD measurement pre- and early post-allotransplant defines very low- and very high-risk ALL patients. Blood. (2015) 125:3501–8. doi: 10.1182/blood-2014-12-615757
23. Brüggemann M, Kotrová M, Knecht H, Bartram J, Boudjogrha M, Bystry V, et al. Standardized next-generation sequencing of immunoglobulin and T-cell receptor gene recombinations for MRD marker identification in acute lymphoblastic leukaemia; a EuroClonality-NGS validation study. Leukemia. (2019) 33:2241–53. doi: 10.1038/s41375-019-0496-7
24. Logan AC, Vashi N, Faham M, Carlton V, Kong K, Buno I, et al. Immunoglobulin and T-cell receptor gene high-throughput sequencing quantifies minimal residual disease in acute lymphoblastic leukemia and predicts post-transplant relapse and survival. Biol Blood Marrow Transplant. (2014) 20:1307–13. doi: 10.1016/j.bbmt.2014.04.018
25. Bartram J, Goulden N, Wright G, Adams S, Brooks T, Edwards D, et al. High throughput sequencing in acute lymphoblastic leukemia reveals clonal architecture of central nervous system and bone marrow compartments. Haematologica. (2018) 103:e110–4. doi: 10.3324/haematol.2017.174987
26. Bahjat M, Guikema JEJ. The complex interplay between DNA injury and repair in enzymatically induced mutagenesis and DNA damage in B lymphocytes. Int J Mol Sci. (2017) 18:1876. doi: 10.3390/ijms18091876
27. Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet. (2014) 46:116–25. doi: 10.1038/ng.2874
28. Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. (2008) 453:110–4. doi: 10.1038/nature06866
29. Malouf C, Ottersbach K. Molecular processes involved in B cell acute lymphoblastic leukaemia. Cell Mol Life Sci. (2018) 75:417–46. doi: 10.1007/s00018-017-2620-z
30. De Moerloose B, Suciu S, Bertrand Y, Mazingue F, Robert A, Uyttebroeck A, et al. Improved outcome with pulses of vincristine and corticosteroids in continuation therapy of children with average risk acute lymphoblastic leukemia (ALL) and lymphoblastic non-Hodgkin lymphoma (NHL): report of the EORTC randomized phase 3 trial 58951. Blood. (2010) 116:36–44. doi: 10.1182/blood-2009-10-247965
31. Domenech C, Suciu S, De Moerloose B, Mazingue F, Plat G, Ferster A, et al. Dexamethasone (6 mg/m2/day) and prednisolone (60 mg/m2/day) were equally effective as induction therapy for childhood acute lymphoblastic leukemia in the EORTC CLG 58951 randomized trial. Haematologica. (2014) 99:1220–7. doi: 10.3324/haematol.2014.103507
32. Wu D, Emerson RO, Sherwood A, Loh ML, Angiolillo A, Howie B, et al. Detection of minimal residual disease in B lymphoblastic leukemia by high-throughput sequencing of IGH. Clin Cancer Res. (2014) 20:4540–8. doi: 10.1158/1078-0432.CCR-13-3231
33. Smith M, Arthur D, Camitta B, Carroll AJ, Crist W, Gaynon P, et al. Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. J Clin Oncol. (1996) 14:18–24. doi: 10.1200/JCO.1996.14.1.18
34. Dastugue N, Suciu S, Plat G, Speleman F, Cavé H, Girard S, et al. Hyperdiploidy with 58-66 chromosomes in childhood B-acute lymphoblastic leukemia is highly curable: 58951 CLG-EORTC results. Blood. (2013) 121:2415–23. doi: 10.1182/blood-2012-06-437681
35. Alves-Pereira CF, de Freitas R, Lopes T, Gardner R, Marta F, Vieira P, et al. Independent recruitment of Igh alleles in V(D)J recombination. Nat Commun. (2014) 5:5623. doi: 10.1038/ncomms6623
36. Theunissen PMJ, van Zessen D, Stubbs AP, Faham M, Zwaan CM, van Dongen JJM, et al. Antigen receptor sequencing of paired bone marrow samples shows homogeneous distribution of acute lymphoblastic leukemia subclones. Haematologica. (2017) 102:1869–77. doi: 10.3324/haematol.2017.171454
37. Bhojwani D, Pei D, Sandlund JT, Jeha S, Ribeiro RC, Rubnitz JE, et al. ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: improved outcome with contemporary therapy. Leukemia. (2012) 26:265–70. doi: 10.1038/leu.2011.227
38. Hübner S, Cazzaniga G, Flohr T, van der Velden VHJ, Konrad M, Pötschger U, et al. High incidence and unique features of antigen receptor gene rearrangements in TEL-AML1-positive leukemias. Leukemia. (2004) 18:84–91. doi: 10.1038/sj.leu.2403182
39. Greaves MF, Wiemels J. Origins of chromosome translocations in childhood leukaemia. Nat Rev Cancer. (2003) 3:639–49. doi: 10.1038/nrc1164
40. Heinäniemi M, Vuorenmaa T, Teppo S, Kaikkonen MU, Bouvy-Liivrand M, Mehtonen J, et al. Transcription-coupled genetic instability marks acute lymphoblastic leukemia structural variation hotspots. eLife. (2016) 5:e13087. doi: 10.7554/eLife.13087
41. Swaminathan S, Klemm L, Park E, Papaemmanuil E, Ford A, Kweon S-M, et al. Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nat Immunol. (2015) 16:766–74. doi: 10.1038/ni.3160
42. Schatz DG, Swanson PC. V(D)J recombination: mechanisms of initiation. Ann Rev Genet. (2011) 45:167–202. doi: 10.1146/annurev-genet-110410-132552
43. Biondi A, Schrappe M, De Lorenzo P, Castor A, Lucchini G, Gandemer V, et al. Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study. Lancet Oncol. (2012) 13:936–45. doi: 10.1016/S1470-2045(12)70377-7
44. Bardini M, Woll PS, Corral L, Luc S, Wittmann L, Ma Z, et al. Clonal variegation and dynamic competition of leukemia-initiating cells in infant acute lymphoblastic leukemia with MLL rearrangement. Leukemia. (2015) 29:38–50. doi: 10.1038/leu.2014.154
45. McHale CM, Smith MT. Prenatal origin of chromosomal translocations in acute childhood leukemia: implications and future directions. Am J Hematol. (2004) 75:254–7. doi: 10.1002/ajh.20030
46. Gawad C, Koh W, Quake SR. Dissecting the clonal origins of childhood acute lymphoblastic leukemia by single-cell genomics. PNAS. (2014) 111:17947–52. doi: 10.1073/pnas.1420822111
47. Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. (2008) 322:1377–80. doi: 10.1126/science.1164266
48. Ribera J, Zamora L, Morgades M, Mallo M, Solanes N, Batlle M, et al. Copy number profiling of adult relapsed B-cell precursor acute lymphoblastic leukemia reveals potential leukemia progression mechanisms. Genes Chromosomes Cancer. (2017) 56:810–20. doi: 10.1002/gcc.22486
49. Ding L-W, Tan K-T, Sun Q-Y, Lao Z-T, Yang H, Jiang N, et al. Clonality and clonal evolution analysis of paediatric all based on B-cell receptor/T-cell receptor rearrangement. Br J Haematol. (2019) 184:829–33. doi: 10.1111/bjh.15179
50. Salson M, Giraud M, Caillault A, Grardel N, Duployez N, Ferret Y, et al. High-throughput sequencing in acute lymphoblastic leukemia: follow-up of minimal residual disease and emergence of new clones. Leuk Res. (2017) 53:1–7. doi: 10.1016/j.leukres.2016.11.009
51. Shin S, Hwang IS, Kim J, Lee K-A, Lee S-T, Choi JR. Detection of immunoglobulin heavy chain gene clonality by next-generation sequencing for minimal residual disease monitoring in B-lymphoblastic leukemia. Ann Lab Med. (2017) 37:331–5. doi: 10.3343/alm.2017.37.4.331
52. Tasian SK, Loh ML, Hunger SP. Childhood acute lymphoblastic leukemia: integrating genomics into therapy. Cancer. (2015) 121:3577–90. doi: 10.1002/cncr.29573
53. Lalonde E, Wertheim G, Li MM. Clinical impact of genomic information in pediatric leukemia. Front Pediatr. (2017) 5:263. doi: 10.3389/fped.2017.00263
54. Szczepañski T, Langerak AW, Wolvers-Tettero IL, Ossenkoppele GJ, Verhoef G, Stul M, et al. Immunoglobulin and T cell receptor gene rearrangement patterns in acute lymphoblastic leukemia are less mature in adults than in children: implications for selection of PCR targets for detection of minimal residual disease. Leukemia. (1998) 12:1081–8. doi: 10.1038/sj.leu.2401071
55. Meleshko AN, Belevtsev MV, Savitskaja TV, Potapnev MP. The incidence of T-cell receptor gene rearrangements in childhood B-lineage acute lymphoblastic leukemia is related to immunophenotype and fusion oncogene expression. Leuk Res. (2006) 30:795–800. doi: 10.1016/j.leukres.2005.11.007
56. Ding L-W, Sun Q-Y, Tan K-T, Chien W, Mayakonda A, Yeoh AEJ, et al. Mutational landscape of pediatric acute lymphoblastic leukemia. Cancer Res. (2017) 77:390–400. doi: 10.1158/0008-5472.CAN-16-1303
57. Müschen M. Rationale for targeting the pre-B-cell receptor signaling pathway in acute lymphoblastic leukemia. Blood. (2015) 125:3688–93. doi: 10.1182/blood-2015-01-567842
58. Brumpt C, Delabesse E, Beldjord K, Davi F, Cayuela JM, Millien C, et al. The incidence of clonal T-cell receptor rearrangements in B-cell precursor acute lymphoblastic leukemia varies with age and genotype. Blood. (2000) 96:2254–61.
59. van der Velden VHJ, Szczepanski T, Wijkhuijs JM, Hart PG, Hoogeveen PG, Hop WCJ, et al. Age-related patterns of immunoglobulin and T-cell receptor gene rearrangements in precursor-B-ALL: implications for detection of minimal residual disease. Leukemia. (2003) 17:1834–44. doi: 10.1038/sj.leu.2403038
Keywords: minimal residual disease (MRD), BCP-ALL, clonal evolution analysis, prognostic factors, high-throughput sequencing (HTS)
Citation: Levy G, Kicinski M, Van der Straeten J, Uyttebroeck A, Ferster A, De Moerloose B, Dresse M-F, Chantrain C, Brichard B and Bakkus M (2022) Immunoglobulin Heavy Chain High-Throughput Sequencing in Pediatric B-Precursor Acute Lymphoblastic Leukemia: Is the Clonality of the Disease at Diagnosis Related to Its Prognosis? Front. Pediatr. 10:874771. doi: 10.3389/fped.2022.874771
Received: 13 February 2022; Accepted: 29 April 2022;
Published: 30 May 2022.
Edited by:
Sarah K. Tasian, Children’s Hospital of Philadelphia, United StatesReviewed by:
Marek Ussowicz, Wrocław Medical University, PolandCopyright © 2022 Levy, Kicinski, Van der Straeten, Uyttebroeck, Ferster, De Moerloose, Dresse, Chantrain, Brichard and Bakkus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriel Levy, Z2EubGV2eUB1Y2xvdXZhaW4uYmU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.