 Yu-Jia Guan
Yu-Jia Guan Yan-Nan Guo3,4*
Yan-Nan Guo3,4*
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 15 April 2022
Sec. Genetics of Common and Rare Diseases
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.860990
This article is part of the Research Topic Inherited Metabolic Diseases in Pediatrics: Clinical and Molecular Features View all 25 articles
Objective: To report a rare case of cystinosis with a novel CTNS pathogenic variant in the Chinese population.
Methods: Retrospective analysis of the clinical manifestations, laboratory results, and gene detection data of a child with cystinosis.
Results: A Chinese Zang ethnic girl could not stand or walk until 3 years old, with additional symptoms including a loss of appetite. Since then, the girl gradually exhibited “X” leg, double wrist joints, a bilateral ankle deformity, and rickets. At the age of 9 years, the girl was hospitalized. Laboratory testing showed that her blood phosphorus, blood calcium and blood potassium levels were significantly decreased. At the same time, the girl's urine glucose and urine protein were positive, although her fasting blood glucose, glycosylated hemoglobin, and 75 g glucose tolerance were not significantly abnormal. Further, blood gas analysis showed metabolic acidosis. These symptoms corresponded to Fanconi syndrome. Gene analysis showed that there was a homozygous pathogenic variant c.140 ≤ 5G > A (p.?) in the CTNS gene, which was a small variation in the intron region. To our knowledge, this is the first report of the rare variant.
Conclusion: Attention should be paid to the differential diagnosis of cystinosis by gene analysis in children whose clinical manifestations include exercise dysplasia, renal damage, or multiple organ damage (including bone, thyroid, etc) and who cannot be firmly diagnosed for the time being.
Cystinosis is an extremely rare autosomal recessive metabolic disease which is caused by the lack of L-cystine transporters on the lysosomal membrane resulted from the pathogenic variant of CTNS. This defect in lysosomal membrane transport leads to cystine accumulation, which eventually causes multiple organ dysfunction, including the eyes, kidneys, nerves, heart, and endocrine gland, amongst others. Until now, the majority of reported Cystinosis cases have occurred in Europe and the United States. The incidence of cystinosis in the general population is within the range of 1:260,000–1:115,000 (1, 2), which varies greatly in different regions. It should be noted that the relevant data in China are still incomplete.
In recent years, some progress has been made in the study of the disease in developed countries. However, relatively few reviews or case reports from China have been published (3–6). Furthermore, cases of cystinosis in children are particularly rare, which may be related to the low incidence of the disease and also the understanding of the disease. Timely findings, accurate diagnosis, and extensive reporting will be helpful to increase awareness of this disease. Meanwhile, early diagnosis and early treatment have an important impact on the prognosis of the disease, and can help to reduce or delay the occurrence of complications and prolong the patient's life. In this paper, the clinical manifestations, laboratory results, and gene detection data of a child with cystinosis diagnosed in West China Second Hospital of Sichuan University were reported (see below). The clinical characteristics, diagnosis and treatment strategies were discussed.
The patient was a 9-year-old girl who was the second child of non-consanguineous parents from the Zang ethnic minority group, Sichuan Province, China. Her mother was not exposed to or used alcohol, tobacco, or drugs during her pregnancy. She was carried full-term and had a normal vaginal delivery, although her birth weight and body length could not be provided by her family. She was breastfed after birth with supplementary foods added when appropriate. At the age of 1, the child began to crawl and sit by herself, which was not something her family paid particular attention to. However, by the age of 3, she could not stand or walk. Her mobility issues were accompanied by a poor diet, which mainly consisted of formula milk. The child's upper limb movement and cognitive and intellectual development were not significantly abnormal compared to children of the same age. Vitamin D and lysine were prescribed for her initial treatment in the outpatient department of a local hospital. At the age of 4 years, she could walk independently and appeared “X” leg. This patient was diagnosed with Fanconi syndrome, which based on renal tubular acidosis, vitamin D deficiency, growth retardation, normocytic anemia and the decreased serum phosphorus and calcium level during her first hospitalization, and diagnosed with bronchitis and hypocalcemia. Then she was treated with sodium dihydrogen phosphate, disodium hydrogen phosphate, sodium citrate + potassium citrate, vitamin D, and calcium.
After her symptoms were alleviated, it was suggested that she continue to take the above drugs after discharge and be followed up with the outpatient service. However, the child neither went to the clinic for follow-up on time, nor took medicine strictly according to the doctor's advice. Her family members admitted that they forgot occasionally and thought it was not such a serious problem. Their poor compliance may be related to distance from hospitals, lack of time, and family cultural background, concept and other reasons.
About 4 years later, at 8 years and 9 months old, the girl experienced fatigue and her hands were bent and claw-like. These symptoms were accompanied by pain, and at this time her family members independently decided to discontinue her medication. One month later, she presented with a paroxysmal dull ache in her hypogastrium, dizziness and headaches, and occasionally felt nauseous when eating greasy foods, soreness when exercised. She had occasional soft coughs, without polydipsia, urorrhagia, and so on.
At the age of 9, she was hospitalized in West China Second University Hospital of Sichuan University. The anthropometric evaluation showed that her stature (bodyweight 18 Kg, height 108 cm) was lower than the 3rd percentile of height and weight for individuals of the same age and sex, and also that her subcutaneous fat was thin. A Pectus carinatum brace was fitted. Upon testing, it was found that her blood phosphorus (1.07 mmol/L), blood calcium (1.72 mmol/L), and blood potassium (3.4 mmol/L) levels were significantly decreased. And it was found that her hemoglobin level (88 g/L) was decreased, while both of the level of Mean Corpuscular Volume (MCV) and the level of mean corpuscular hemoglobin (MCH) were within the normal range. This indicated that the patient can be diagnosed with normocytic anemia.
Meanwhile, laboratory tests showed a decreased Vitamin D (both 25-OH-D and 1.25-OH-D) level (9.6 ng/ml), an increased alkaline phosphatase level (575 U/L), significantly increased blood urea nitrogen level (6.98 mmol/L) and creatine level (163 μmol/L). Urine sugar (3 +) and urine protein (2 +) were positive. Her levels of free carnitine (6.1 umol/L), citrulline (44.86 umol/L), glucose (75 g) tolerance, glycosylated hemoglobin, fasting blood glucose, and thyroid function were normal. Blood gas analysis showed metabolic acidosis and anion gap. Renal ultrasound image (Figure 1) showed slightly enhanced parenchymal echo in both kidneys.
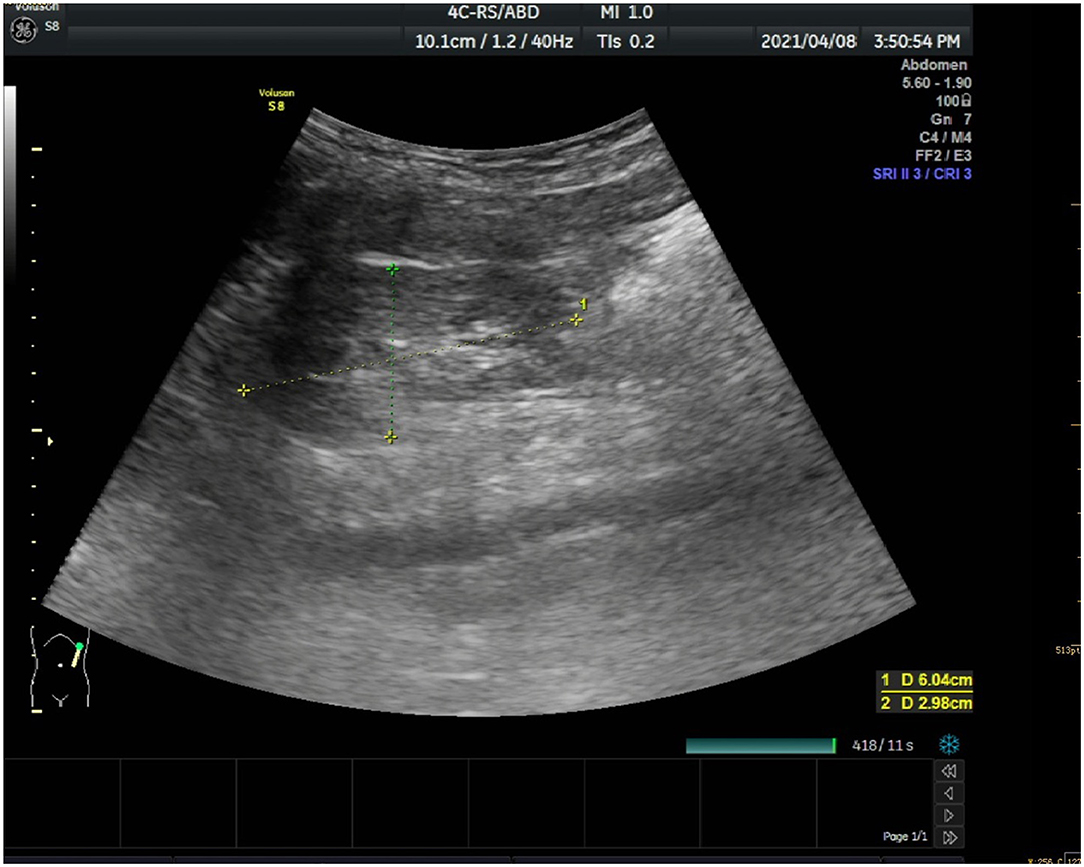
Figure 1. Renal ultrasound image.
The child's clinical symptoms included abnormal motor development for 6 years, with bone deformity, “X” legs, chicken chest, double wrist joints, bilateral ankle joint deformity, and other changes in her left bone and distal radius, ulna distal osteoporosis, including the decreased bone mineral density detected by bone densitometry of child via Quantitative Ultrasound Bone Densitometry (QUS), and left radius and distal ulna rickets. The child had rickets and hypophosphatemia in the past. A diagnosis of Fanconi syndrome was the preliminary clinical consideration. In addition, according to Schwartz formula, the Epidermal Growth Factor Receptor (eGFR) of the patient was 20.17 mL/(min 1.73 m2), indicating that the child was in chronic kidney diseases (stage IV) and did not meet the criteria for dialysis, so dialysis treatment was not performed for the time being. At the same time, we informed the patient's family that the patient was about to enter chronic kidney diseases (stage V), and preparations before dialysis should be made, including indwelling central venous catheterization for hemodialysis and peritoneal dialysis.
The patient suffered from renal tubular acidosis, hypokalemia, hypocalcemia and hypophosphatemia since childhood. The examination after admission showed that urine sugar (3 +) and urine protein (2 +) were positive, while glucose (75 g) tolerance, glycosylated hemoglobin and fasting blood glucose were normal. In addition, α1-microglobulin, β2-microglobulin and other proteins in urine that reflect the reabsorption function of renal tubules were increased. Therefore, the patient was considered to have renal tubular injury, which can lead to electrolyte disturbance, renal diabetes, and partial tubular proteinuria.
Meanwhile, considering the renal tubular injury, the decrease of serum calcium/phosphorus concentration/level and the increase of parathyroxine levels (945.8 pg/ml), the secondary hyperparathyropathy could be diagnosed. Furthermore, the girl had abdominal pain for more than 3 months prior to hospitalization. A CT scan showed slightly blurred mesenteric fat space, multiple enlarged lymph nodes in the abdominal cavity were identified, thus mesenteric lymphadenitis was considered.
During hospitalization, the girl appeared tetany repeatedly, manifested as flexion and rigidity of the limbs as well as chicken claw-like hands accompanied by pain, and the lowest blood calcium level was 1.68 mmol/L. Calcium carbonate was then supplemented intravenously and oral calcium zinc gluconate and ossified triol were added. As citrate would combine with free calcium ions, which could further decrease blood calcium levels, the administration of citrate was stopped.
The girl underwent ophthalmologic examination in West China Fourth Hospital of Sichuan University. The conjunctiva, cornea, iris, lens, retina, intraocular pressure, and diopter were normal, no binocular corneal crystalline was found, and there was no visual field defect.
On the premise of informed consent, the whole exome sequencing (WES) of this girl was conducted. We decided to perform WES analysis because the initial onset age of the patient was early, accompanied by multiple organ system damage, and the treatment effect of oral sodium and potassium citrate mixture for renal tubular acidosis was not good. Meanwhile, the renal function of this patient showed progressive decline. The progression of renal tubular dysfunction, followed by glomerular involvement and renal decompensation, led us to strongly suspect that the patient had a mono-genetic inherited related renal tubule dysfunction.
Gene analysis showed that there was a homozygous pathogenic variant c.140 ≤ 5G > A (p.?) in the patient's CTNS gene (Figure 2). This was a small variation in the intron region, found for the first time. Notably, such a variant has not been reported in the literature and is rare in the population. Combined with the results of genetic examination, the diagnosis of cystinosis was made.
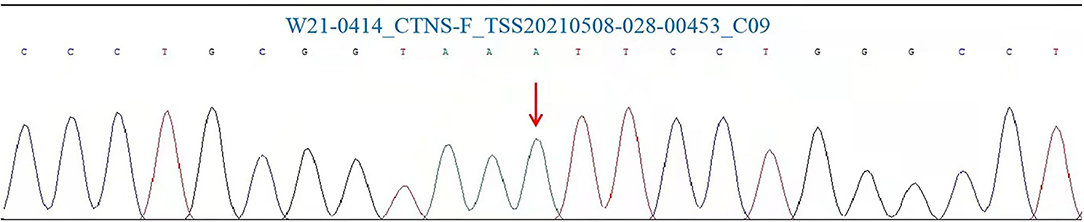
Figure 2. CTNS mutation Sanger analysis results for the patient involved in this study. Polymerase chain reaction (PCR) and Sanger was performed in our test.
Although the child was in a serious condition, her parents asked for her to be discharged after 6 days of hospitalization. Research shows that hypophosphatemia and hypocalcemia in Fanconi syndrome and proximal renal tubular acidosis can be exacerbated by vitamin D deficiency, and the resulting tetany and obvious bone pain. And the above clinical symptoms can be effectively alleviated by the treatment with sodium hydrogen phosphate and sodium phosphate disodium buffer solution and active vitamin D.
After discharge, the parents were guided to supervise the girl to use drugs according to doctor's prescriptions (to take orally sodium dihydrogen phosphate, disodium hydrogen phosphate, lysine inositol vitamin B12, lysine calcium hydrogen phosphate tablets, calcitriol soft capsules, ferrous succinate tablets and vitamin C, and to recheck regularly).
The girl was asked to limit the intake of high protein foods, and return to the hospital if there was any discomfort. Twenty-eight days after the discharge, her family received the gene report results and were advised to carry out the related gene detection. Unfortunately, her family refused to carry out genetic testing. We used network follow-up to remind her family members to take her to the local hospital for urine review to monitor kidney function, but the patient did not follow up strictly according to the doctor's advice.
Up to date, most of the reported cases of cystinosis have occurred in Europe and the United States, with much less cases of cystinosis reported in Asia. Specifically, one case in Japan (7), 6 cases in Thailand (4 families involved) (8), 2 cases in India (9), and 21 cases in China have been reported (Table 1) (3, 5, 6, 10–13). The Chinese cases included 10 Mainland families (including 13 patients with CTNS homozygous mistranslation variant N323K, c.681 G > A(p.E227E), c.477C > G (p.S159R), c.274C > T (p.Q92X), c.680A > T (p.E227V) and CTNS pathogenic variant of (IVS6+1, del G and IVS8-1, and del GT), or with a deletion of c.18_21del GACT, (p.T7FfsX7)), and 5 Taiwanese families (including 8 patients with CTNS homozygous mistranslation variant N323K, 753G > A(W138X), exon 11 (IVS11+2T > C), 1178A-G (K280R) or mutation 57-kb deletion cystinosis).
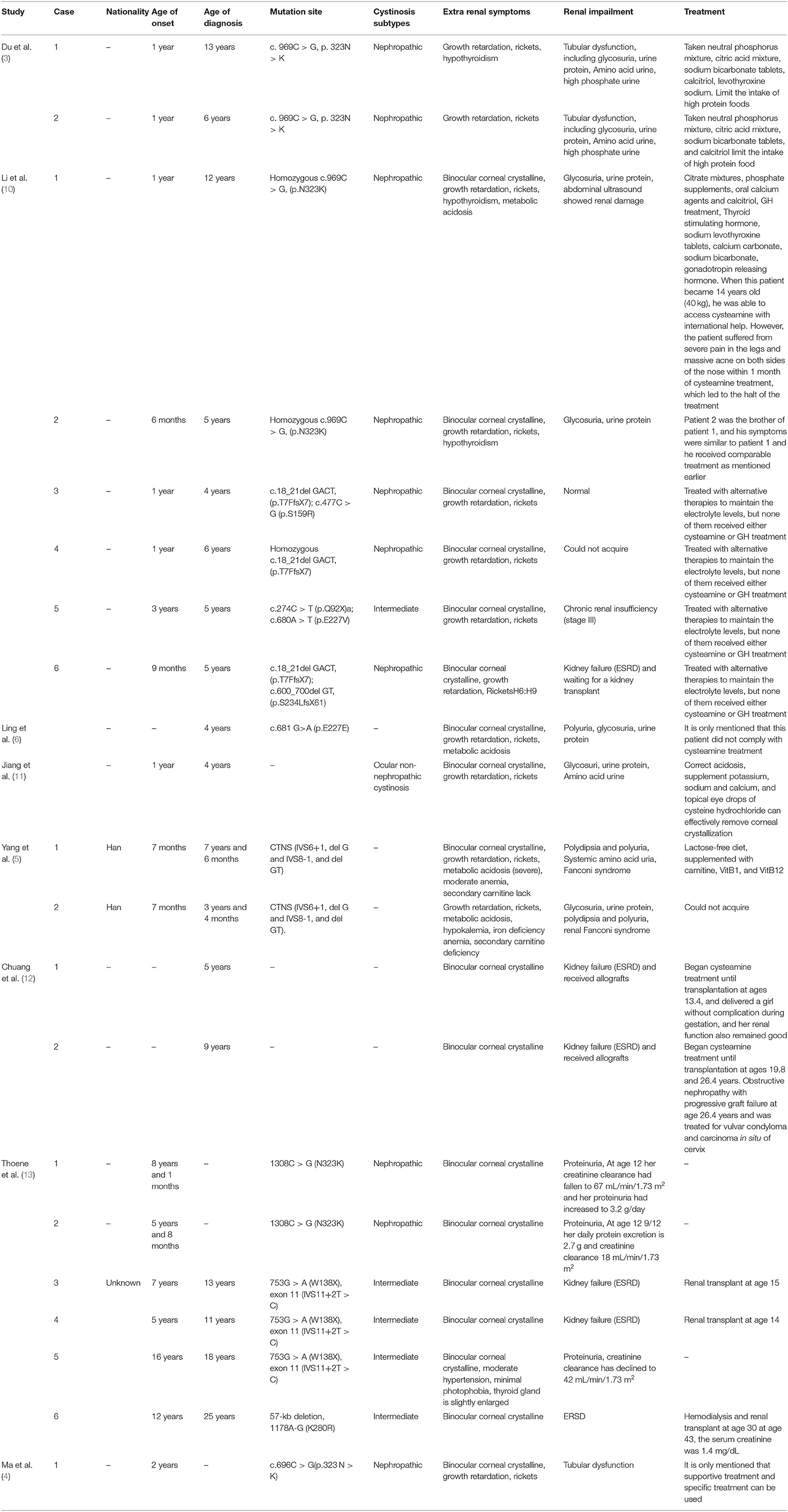
Table 1. Clinical and molecular characteristics of Chinese patients with cystinosis.
At present, the known types of gene pathogenic variant include insertion, deletion, repetition, translocation, point variant, splice site variant, promoter variant, and genome rearrangement of CTNS gene sites. Here we report a new homozygous variant of the CTNS gene. The pathogenic variant site was lntron4 position on chr17:3550821 chromosome, and the protein level of reference transcript was NM 004937.3: c.140+5G > A (p.?). The variation is a small variation in the intron region, which has not been reported in the literature as this variant is rare in the population. The variation cannot be found in the ESP6500siv2 ALL, 1000g2015aug ALL, EXAC, gnomAD, and dbSNP147 databases. Unfortunately, the family members of the child refused to undergo genetic testing, and as such, we could not determine whether other members of the family had similar variations.
Also, among the cases reported in China, only Yong-jia Yang's report clarified ethnic groups of cystinosis patients, in which a Chinese Han family was affected.
Compared with adults, there are very few reports on cystinosis in children in China. However, in recent years, scholars are increasingly paying attention to cystinosis in children. Based on the duration of presented symptoms and the severity of renal involvement, Cystinosis can be divided into infant type (also known as kidney type), juvenile type (also known as intermediate type), and ophthalmopathy type, of which infant type is the most common and most serious, typically characterized by kidney involvement as the first symptom (14). In our report, the patient belongs to infant type, manifested as involvement of multiple organs such as kidney, bone, and thyroid. The girl began to develop symptoms at the age of 3 and was first diagnosed with Fanconi syndrome at the age of 4. At the age of 9, she was hospitalized due to the aggravation of the symptoms. At this time, following gene detection, she was diagnosed with cystinosis and chronic kidney disease. It took 5 years from the initial visit to the final diagnosis for this case. This may be related to the low medical cooperation of the patient's family members, characterized by irregular medical treatment, poor medication compliance, etc. There are also other factors, including the rarity of the cases, economic conditions, limit of diagnosis, and treatment technologies as well as the concept of medical behavior of the patient and her family members. In recent years, with the development of economy and the progress of diagnosis and treatment technologies, gene detection technology has gradually become popularized in China, which has greatly enriched the means of diagnosis and treatment and improved the level of treatment.
At present, on the premise of combining clinical manifestations and laboratory results, there are three methods to assist in the diagnosis of suspected cystinosis patients: (1) Using mass spectrometer liquid chromatography to detect the level of cystine in blood leukocytes. This method has high sensitivity and specificity, and can monitor the therapeutic effect. However, as the requirements for instruments and testers are high, it cannot be easily conducted in a clinic. (2) Most patients can be diagnosed by gene detection, although the price is relatively high. (3) Using a slit lamp to detect characteristic corneal cystine crystallization is simple and economical; however, the scope of application is narrow: it is only suitable for patients with eye symptoms and cannot monitor the therapeutic effect.
Therefore, the corresponding medical testing methods can be selected based on the local medical conditions and the clinical manifestation type of the patient in question. At present, some cases of cystinosis in China have been analyzed by gene analysis, with the number of such cases increasing in recent years (3, 5, 6, 10–13). Gene diagnosis is beneficial to the diagnosis and analysis of cystinosis, and it is also an area for development in the future (15).
Some studies have pointed out that, the cases diagnosed may only be part of the actual cases because of the difficulty of diagnosis, such that some patients may not be able to be diagnosed (16). If patients with cystinosis cannot be diagnosed in time, they will not be treated in time, meanwhile the genetic information and clinical data of this population will be missing. From this case, it can be seen that the treatment compliance of the child and her families is not high. Thus, medical and nursing staff need to work to improve the treatment compliance of children and their families.
Health outcomes can be improved through health education and the improvement of drug research and treatment methods, such as developing sustained-release capsules to reduce the frequency of drug taking (17), and informing children and their families of the serious consequences of drug withdrawal through real cases examples.
At present, mercaptamine bitartrate, a drug that can specifically reduce cystine in lysosomes, is the preferred treatment for nephropathic cystinosis. The drug was transported to lysozyme by lysosomal membrane PQLC2 vector by forming cysteamine cysteine binding molecule with cystine in lysozyme. Early medication can effectively improve the growth and development, delay the occurrence of end-stage renal disease, but cannot prevent the progress of the disease. A new cysteamine preparation with delayed release simplifies the administration schedule, but still cannot cure cystinosis disease. Mercaptamine bitartrate is widely used in developed countries, but has not yet been introduced into China, which limits our research on this disease.
In the future, therapeutic research may be carried out based on CTNS-carrying stem cell transplantation, which has been successfully carried out in mouse model. Hematopoietic stem cell transplantation and gene editing technology are possible choices to cure the diseases (18).
As our understanding has developed, cystinosis has gradually become a treatable rare disease. Going forward, it is very important to improve the awareness by timely reporting of the disease and to adopt the behavior of actively seeking medical treatment. Early detection and early treatment can reduce and delay the occurrence of complications and improve the prognosis of patients with cystinosis.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This article was supported by the fund of Science and Technology Bureau of Sichuan Province (Grant No. 2019YFS0240).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Dr. Wen Deng, from The University of Hong Kong, for editing the English text of a draft of this article.
1. Hutchesson AC, Bundey S, Preece MA, Hall SK, Green A. A comparison of disease and gene frequencies of inborn errors of metabolism among different ethnic groups in the West Midlands,UK. J Med Genet. (1998) 35:366–70.
2. Elmonem MA, Veys KR, Soliman NA, Van Dyck M, Van Den Heuvel LP, Levtchenko E. Cystinosis:a review. Orphanet J Rare Dis. (2016) 11:47. doi: 10.1186/s13023-016-0426-y
3. Du J, Pang R, Jiang Y, Wang O, Li M, Xing XP, et al. Clinical manifestations and CTNS gene mutation in two patients with cystinosis. Chin J Osteoporosis Bone Miner Res. (2017) 10:469–73. doi: 10.3969/j.issn.1674-2591.2017.05.007
4. Ma YY, Shen YJ, Zhou L, Liu YP, Li DX, Ding Y, et al. CTNS gene mutation leads to cysteine nephropathy combined with corneal crystal in young child. J Clin Pediatr. (2016) 34:783–6. doi: 10.3969/j.issn.1000-3606.2016.10.017
5. Yang YJ, Hu Y, Zhao R, He XY, Zhao L, Tu M, et al. First report of CTNS mutations in a Chinese family with infantile cystinosis. Sci World J. (2015) 2015:309410. doi: 10.1155/2015/309410
6. Ling C, Liu XR, Chen Z, Jiang YP, Fan JF, Meng Q, et al. Corneal cystine crystals in cystinosis. Arch Dis Childh. (2017) 102:185. doi: 10.1136/archdischild-2016-312456
7. Okami T, Nakajima M, Higashino H, Aoki T. Ocular manifestations in a case of infantile cystinosis. Nippon Ganka Gakkai Zasshi. (1992) 96:1341.
8. Yeetong P, Tongkobpetch S, Kingwatanakul P, Deekajorndech T, Bernardini IM, Suphapeetiporn K, et al. Two novel CTNS mutations in cystinosis patients in Thailand. Gene. (2012) 499:323–5. doi: 10.1016/j.gene.2012.03.047
9. Tang S, Danda S, Zoleikhaeian M, Simon M, Huang T. An Indian boy with nephropathic cystinosis: a case report and molecular analysis of CTNS mutation. Genet Test Mol Biomark. (2009) 13:435–8. doi: 10.1089/gtmb.2008.0156
10. Li XQ, Wu D, Liang XJ, Li WJ, Liu M, Cao BY, et al. The diagnosis of cystinosis in patients reveals new CTNS gene mutations in the Chinese population. J Pediatr Endocrinol Metab. (2019) 32:375–82. doi: 10.1515/jpem-2018-0263
11. Jiang JJ, Bai DY, Yu G, Wu Q. Fanconi syndrome with corneal lesions secondary to cystine disease: a case report. Natl Med J China. (2014) 94:3357–8. doi: 10.3760/cma.j.issn.0376-2491.2014.42.019
12. Chuang YW, Wen MC, Wu MJ, Shu KH, Cheng CH, Yu TM, et al. Follow-up and treatment of renal transplantation with nephropathic cystinosis in central Taiwan. Transplant Proc. (2012) 44:80–2. doi: 10.1016/j.transproceed.2011.12.071
13. Thoene J, Lemons R, Anikster Y, Mullet J, Paelicke K, Lucero C, et al. Mutations of CTNS causing intermediate cystinosis. Mol Genet Metab. (1999) 67:283–93.
14. Harrison F, Yeagy BA, Rocca CJ, Kohn DB, Salomon DR, Cherqui S. Hematopoietic stem cell gene therapy for the multisystemic lysosomal storage disorder cystinosis. Mol Ther. (2013) 21:433–44. doi: 10.1038/mt.2012.214
15. Jiang XY, Rong LP. Progres of diagnosis and treatment for Gitelman syndrome and cystinosis. Chin J Appl Clin Pediatr. (2018) 33:1296–300. doi: 10.3760/cma.j.issn.2095-428X.2018.17.005
16. Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med. (2002) 347:111–21. doi: 10.1056/NEJMra020552
17. Dohil R, Fidler M, Gangoiti JA, Kaskel F, Schneider JA, Barshop BA. Twice-daily cysteamine bitartrate therapy for children with cystinosis. J Pediatr. (2010) 156:71–5. doi: 10.1016/j.jpeds.2009.07.016
Keywords: cystinosis, CTNS gene mutation, Fanconi syndrome, renal tubular acidosis, genetics
Citation: Guan Y-J, Guo Y-N, Peng W-T and Liu L-L (2022) Case Report: Cystinosis in a Chinese Child With a Novel CTNS Pathogenic Variant. Front. Pediatr. 10:860990. doi: 10.3389/fped.2022.860990
Received: 24 January 2022; Accepted: 21 March 2022;
Published: 15 April 2022.
Edited by:
Hui Xiong, Peking University First Hospital, ChinaReviewed by:
Roberto Chimenz, University of Messina, ItalyCopyright © 2022 Guan, Guo, Peng and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan-Nan Guo, dGhlcmVzYV9ndW9AdG9tLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.