 Aviva Eliyahu1,2Ortal Barel3,4Lior Greenbaum1,2,5Gal Zaks Hoffer2,6Yael Goldberg2,6Annick Raas-Rothschild2,7Amihood Singer8Ifat Bar-Joseph3,4Vered Kunik9Elisheva Javasky3,4
Aviva Eliyahu1,2Ortal Barel3,4Lior Greenbaum1,2,5Gal Zaks Hoffer2,6Yael Goldberg2,6Annick Raas-Rothschild2,7Amihood Singer8Ifat Bar-Joseph3,4Vered Kunik9Elisheva Javasky3,4 Orna Staretz-Chacham10,11Naomi Pode-Shakked2,12,13Lily Bazak6,14,15Noa Ruhrman-Shahar2,6Elon Pras1,2Moshe Frydman1,2Mordechai Shohat2,3
Orna Staretz-Chacham10,11Naomi Pode-Shakked2,12,13Lily Bazak6,14,15Noa Ruhrman-Shahar2,6Elon Pras1,2Moshe Frydman1,2Mordechai Shohat2,3 Ben Pode-Shakked2,7,13*
Ben Pode-Shakked2,7,13*- 1The Danek Gertner Institute of Human Genetics, Sheba Medical Center, Ramat Gan, Israel
- 2Sackler Faculty of Medicine, Tel-Aviv University, Tel-Aviv, Israel
- 3The Genomic Unit, Sheba Cancer Research Center, Sheba Medical Center, Ramat Gan, Israel
- 4The Wohl Institute for Translational Medicine and Cancer Research Center, Sheba Medical Center, Ramat Gan, Israel
- 5The Joseph Sagol Neusroscience Center, Sheba Medical Center, Ramat Gan, Israel
- 6The Raphael Recanati Genetics Institute, Rabin Medical Center - Beilinson Hospital, Petah Tikva, Israel
- 7The Institute for Rare Diseases, Edmond and Lily Safra Children's Hospital, Sheba Medical Center, Ramat Gan, Israel
- 8Department of Community Genetics, Public Health Services, Ministry of Health, Jerusalem, Israel
- 9Bioinformatics Consulting, Maas, Israel
- 10Metabolic Clinic, Soroka Medical Center, Be'er Sheva, Israel
- 11Faculty of Health Sciences, Ben-Gurion University, Be'er Sheva, Israel
- 12Department of Pediatrics, Edmond and Lily Safra Children's Hospital, Sheba Medical Center, Ramat Gan, Israel
- 13The Talpiot Medical Leadership Program, Sheba Medical Center, Ramat Gan, Israel
- 14Mina and Everard Goodman Faculty of Life Science, Bar Ilan University, Ramat Gan, Israel
- 15Felsenstein Medical Research Center, Rabin Medical Center, Petah Tikva, Israel
The role of lysine methyltransferases (KMTs) and demethylases (KDMs) in the regulation of chromatin modification is well-established. Recently, deleterious heterozygous variants in KMT5B were implicated in individuals with intellectual disability (ID) and/or autism spectrum disorder. We describe three unrelated patients with global developmental delay (GDD) or ID, macrocephaly and additional features. Using whole exome sequencing, each of the probands was found to harbor a distinct de novo heterozygous disease-causing variant in KMT5B: c.541C > G (p.His181Asp); c.833A > T (p.Asn278Ile); or c.391_394delAAAG (p.Lys131GlufsTer6). We discuss herein their clinical presentations, and compare them to those of previously reported patients. Furthermore, using a three-dimensional computational model of the KMT5B protein, we demonstrate the predicted structural effects of the two missense variants. Our findings support the role of de novo missense and nonsense variants in KMT5B-associated GDD/ID, and suggest that this gene should be considered in the differential diagnosis of neurodevelopmental disorders accompanied by macrocephaly and/or overgrowth.
Introduction
Methyltransferases (KMTs) and demethylases (KDMs) play an important role in the regulation of chromatin modification (1, 2). Pathogenic variants in several of these genes have been reported in autosomal-dominant or X-linked neurodevelopmental disorders. Most recently, heterozygous disease-causing variants in the Lysine Methyltransferase 5B (KMT5B) gene, have been described in individuals with neurodevelopmental delay, with or without seizures and/or autism spectrum disorder (ASD) (MIM #617788).
Initially, KMT5B (also termed SUV420H1) was suggested as a candidate gene associated with ASD, using computational modeling (3). Only in 2017, in a cohort of ~11,700 patients with neurodevelopmental delay, Stessman et al. found deleterious heterozygous variants in KMT5B in seven probands (4). In addition to intellectual disability (ID) (7/7), other features included ASD (5/6), febrile seizures (3/5) and attention deficit (3/7). Of these, three patients had previously been reported as part of a cohort of 2,500 ASD cases (5). Since then, several additional individuals with varying degrees of ID harboring de novo heterozygous KMT5B variants were reported (6, 7).
Functionally, the role of KMT5B in habituation learning was demonstrated in a Drosophila model (4). Most recently, two mouse models for KMT5B were reported. Using a comprehensive neurodevelopmental test battery, Wickramasekara et al. showed differences with regard to anxiety, depression, fear and extinction learning in mice haploinsufficient for KMT5B compared to controls, as well as differential effects by sex (8, 9). Wang et al. knocked down Kmt5b in the mouse prefrontal cortex and demonstrated social deficits, disruption of glutamatergic synaptic transmission as well as upregulation of genes involved in cellular stress response and ubiquitin-dependent protein degradation (10).
We describe herein clinical and molecular features of three unrelated patients with de novo disease-causing variants in Kmt5b, demonstrate the missense variants' predicted effects in a three dimensional (3D) computational model of the protein, and compare the patients' phenotypic features to those of previously-described patients.
Materials and Methods
Following parental written informed consent, blood samples from the probands and their parents were obtained for trio Whole exome sequencing (WES). The study was approved by the Institutional Review Board of Sheba Medical Center. WES analysis was performed at the Genomic Unit of the Sheba Cancer Research Center (Probands A,B). These two individuals were included in our previous publication, describing our experience with 280 individuals with neurodevelopmental disorders or multiple congenital anomalies (11). However, the current study describes their clinical phenotype in detail. Proband C was not reported previously, and WES was performed at the Raphael Recanati Genetic Institute at Beilinson Medical Center, following IRB approval.
Whole Exome Sequencing
Trio-based Whole Exome Sequencing (WES) was performed on genomic DNA at the Genomic Unit, Sheba Medical Center (for families A and B), or at the Raphael Recanati Genetics Institute (for family C), using the Twist Human Core Exome Plus Kit (Twist Bioscience, San Francisco, CA, USA). Detailed methods and technologies used for sequencing, variant calling and interpretation are described in the Supplementary Material.
3D Modeling of Wild-Type KMT5B and H181D and N278I Variants
The 3D structural models of wild-type (WT) and mutated KMT5B structures were constructed using the SWISS-MODEL protein structure homology-modeling server (12), using the PDB structure 5cpr as the template for structure prediction. The same 3D modeling algorithm was used to construct WT and mutated structures to avoid differences that may arise due to the modeling process.
Structural Analysis
Discovery Studio® Visualizer (version 16.1.0.15350, BIOVIA, Dassault Systèmes Discovery Studio Modeling Environment, Release 4) was used to calculate intra-structure hydrogen bonds and the distances between atoms.
Results
Clinical Characteristics
Proband A presented at 12 months of age due to global developmental delay (GDD). The first child born to non-consanguineous healthy parents of mixed Jewish descent, he was born after an uneventful pregnancy and delivery at term (birth weight, 3,212 g). At 3 months of age, neurological and ophthalmological evaluation was pursued due to lack of eye contact. Delay in acquisition of developmental milestones was noted: he raised his head at 5.5 months, rolled over at 7 months and began crawling at 9 months of age. He showed profound GDD and macrocephaly. Parents noted episodes of unexplained laughter. At presentation at the age of 12 months, he was planned to undergo surgical repair of lateral strabismus. Upon physical examination, a broad forehead, strabismus, short neck and mild hypertelorism were noted. Weight was 10 kg (40th percentile) and head circumference was 49.2 cm (>97th percentile). Electroencephalogram (EEG), brain ultrasound and magnetic resonance imaging (MRI) were all intact.
Proband B presented at 2.5 years of age, due to GDD and relative macrocephaly. He was the first child of healthy, non-consanguineous Ashkenazi-Jewish parents, and born following a spontaneous, uneventful pregnancy, via post-term delivery (birth weight, 3,800 g). During infancy, brain ultrasound was performed due to large head circumference, and was normal. At 12 months he began passing loose stools, and was diagnosed with lactose intolerance. Ophthalmological evaluation revealed astigmatism and hypermetropia, and he began wearing glasses at 1.5 years of age. Delayed requisition of developmental milestones was evident, as he began walking at 22 months, and said his first words at 26 months. Upon evaluation (30 months), he had severe GDD, with an overall developmental quotient (DQ) of 50. Seizures were not documented. Upon physical examination, weight was 12.1 kg (30th percentile), height was 83.4 cm (10th percentile) and head circumference was 52 cm (95th percentile). Broad forehead, conical fingers, hypotonia and joint hypermobility were noted.
Proband C was evaluated at 35 years of age due to ID, epilepsy and overgrowth. The fourth of five children born to non-consanguineous healthy parents of Arab-Muslim descent, he was born after an uneventful pregnancy and delivery at term (birth weight, 4,500 g). Delay in acquisition of developmental milestones was observed: He walked at the age of 3 years, spoke first words at the age of 4 years. At the age of 8 years he experienced his first generalized seizure, followed by a second and last seizure, with no additional events under anticonvulsant therapy. His growth parameters were reported as above the average, however starting at the age of 10 years he began to grow at an accelerated rate. Upon physical examination at 35 years, morbid obesity, occipital protrusion and upsweeping of frontal hair were noticed, as well as large hands with long and lean fingers. Height was 205 cm (>97th percentile), weight was 150 kg (>97th percentile) and head circumference was 62 cm (>97th percentile). Brain CT was considered normal. Brain MRI demonstrated a 6 x 8 mm non-functioning pituitary lesion which was considered to be an incidental finding without clinical relevance. Hormonal profiling showed decreased testosterone levels.
For all three patients, chromosomal microarray analysis (CMA) and maternal molecular analysis for Fragile X (FRAX) syndrome were normal. For proband B, targeted Sanger sequencing of PTEN was normal.
Molecular Analysis—Whole Exome Sequencing
In order to reach a molecular diagnosis, trio-WES was pursued for each of the probands. Each of the three probands presented a distinct, de-novo heterozygous variant in KMT5B (NM_017635.5). Two were of missense type: c.541C > G (p.His181Asp) in proband A, and c.833A > T (p.Asn278Ile) in proband B. In proband C, we identified a c.391_394delAAAG (p.Lys131GlufsTer6) frameshift variant. For probands A and B, harboring the missense variants, the affected amino acid residues are highly conserved throughout evolution (Figure 1), are extremely rare (not found in public databases, including gnomAD, nor in the Sheba Medical Center in-house database) and predicted as damaging/pathogenic by several software (including PolyPhen-2, MutationTaster, FATHMM and SIFT). With regard to the missense variants, the gene pLI and Z score are 1 and 2.79, respectively. The CADD scores for the p.His181Asp and the p.Asn278Ile missense variants were 28.7 and 31, respectively. The p.His181Asp variant was classified as likely-pathogenic (meeting the ACMG criteria: PM2, PP3, PS2), and the two others as pathogenic (ACMG criteria: PM2, PP3, PS2 for the p.Asn278Ile variant and PVS1, PM2, PS2 for p.Lys131Glufs). The p.Asn278Ile is likely to be deleterious to the enzyme activity, as it is located in the SET domain, close to the S-adenosyl-L-methionine and Zinc binding region of the catalytic domain, which are crucial for its function, and in which two pathogenic variants have been previously reported (Figure 1). Table 1 summarizes the disease-causing variants in previously-reported patients, and those described herein.
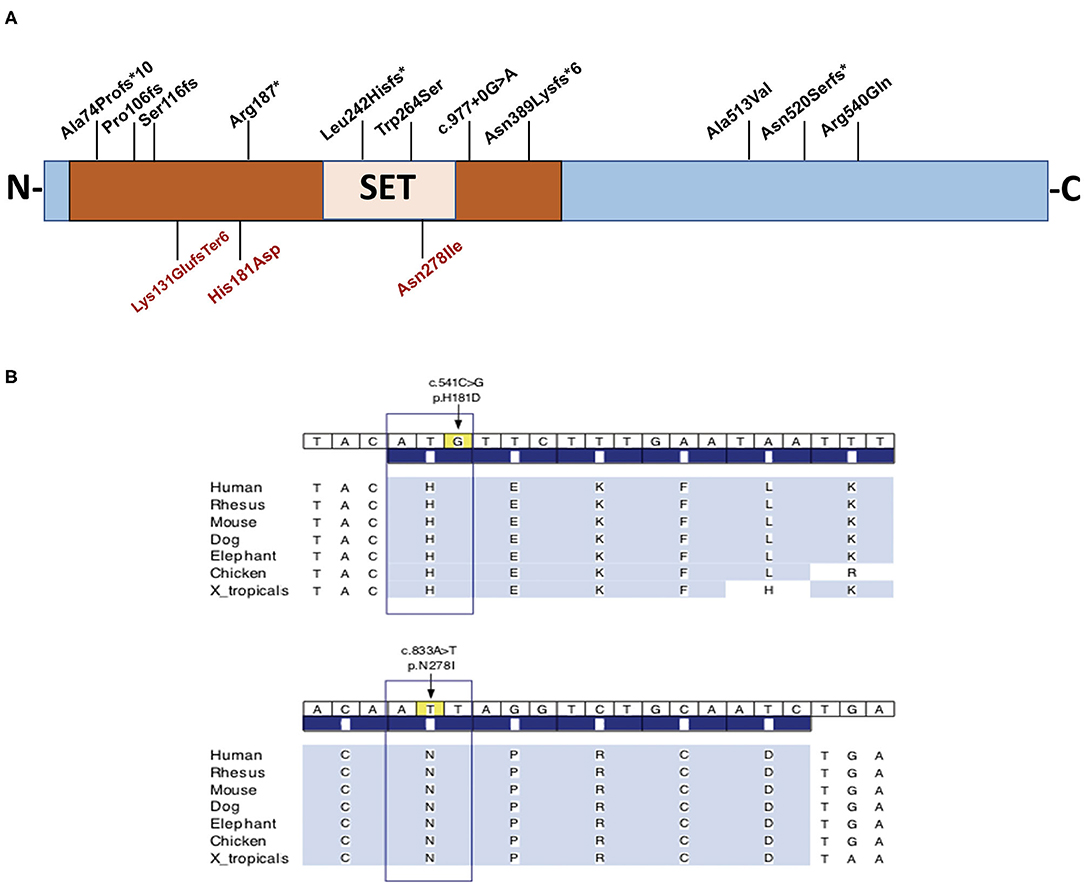
Figure 1. (A) Schematic representation of the full length KMT5B protein and the location of disease-causing variants in three unrelated patients, and all previously-reported patients for whom phenotypic features were available (n = 14). The variants identified in the three individuals in this report appear on the bottom. (B) Conservation of amino acids H181 and N278 throughout evolution. Brown box designates the functional domain of KMT5B as a histone-lysine N-methyltransferase.
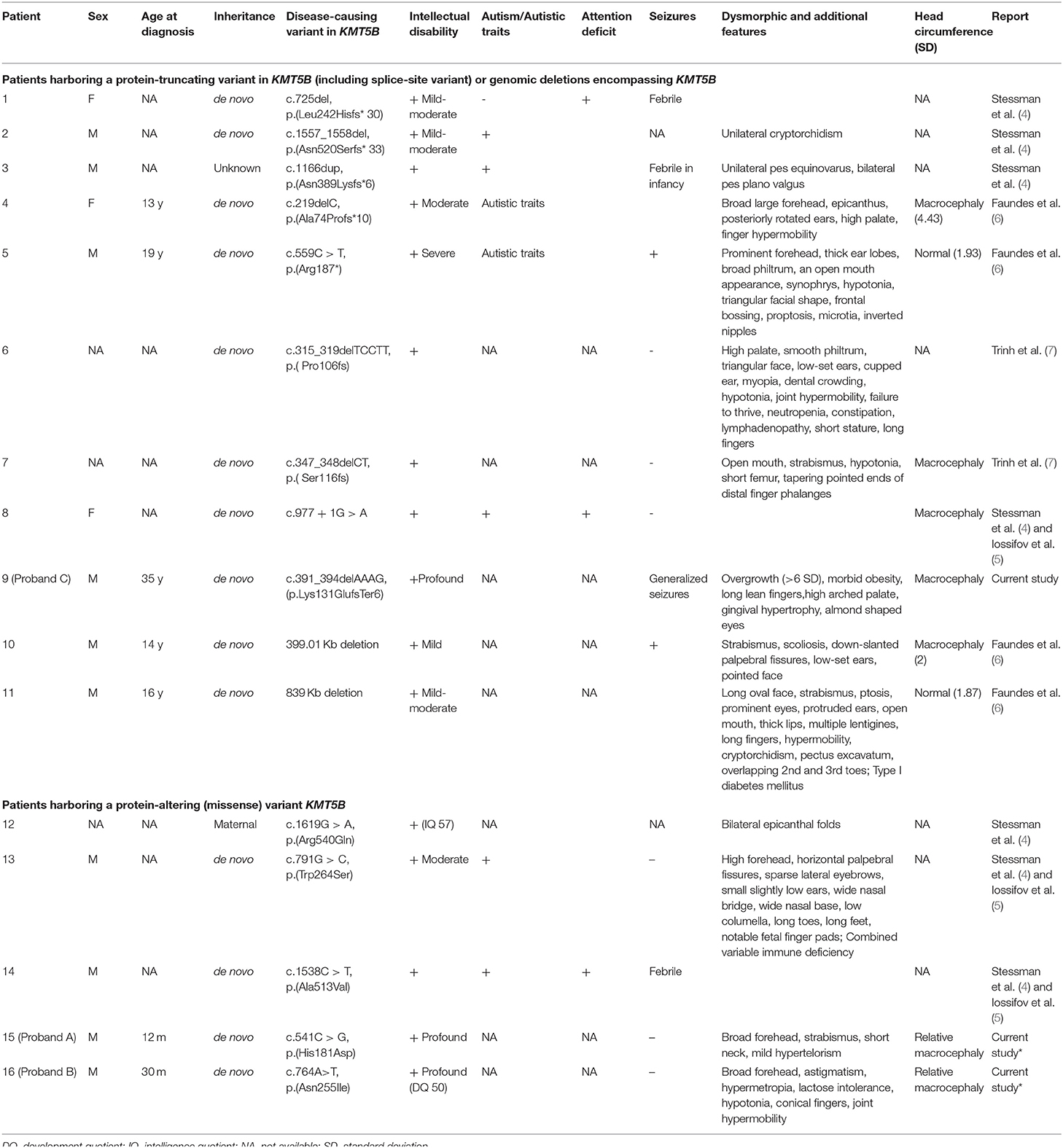
Table 1. Clinical and molecular characteristics of individuals harboring disease-causing variants in KMT5B or deletions encompassing KMT5B.
Residue H181 Forms Two Hydrogen Bonds and Residue N278 Forms Five Hydrogen Bonds in the Wild-Type KMT5B Structure
We analyzed the spatial location of residues H181 and N278 (identified missense variant sites) and the hydrogen bonds they form. Figure 2A shows the spatial location of residues H181 and N278 (ball and stick), the residues with they form hydrogen bonds (stick) as well as the residues that are with 5Å distance (stick) within wild-type KMT5B (cyan, solid ribbon). The hydrogen bonds formed by N278 and H181 in wild-type KMT5B are presented in Figures 2B,C, respectively.
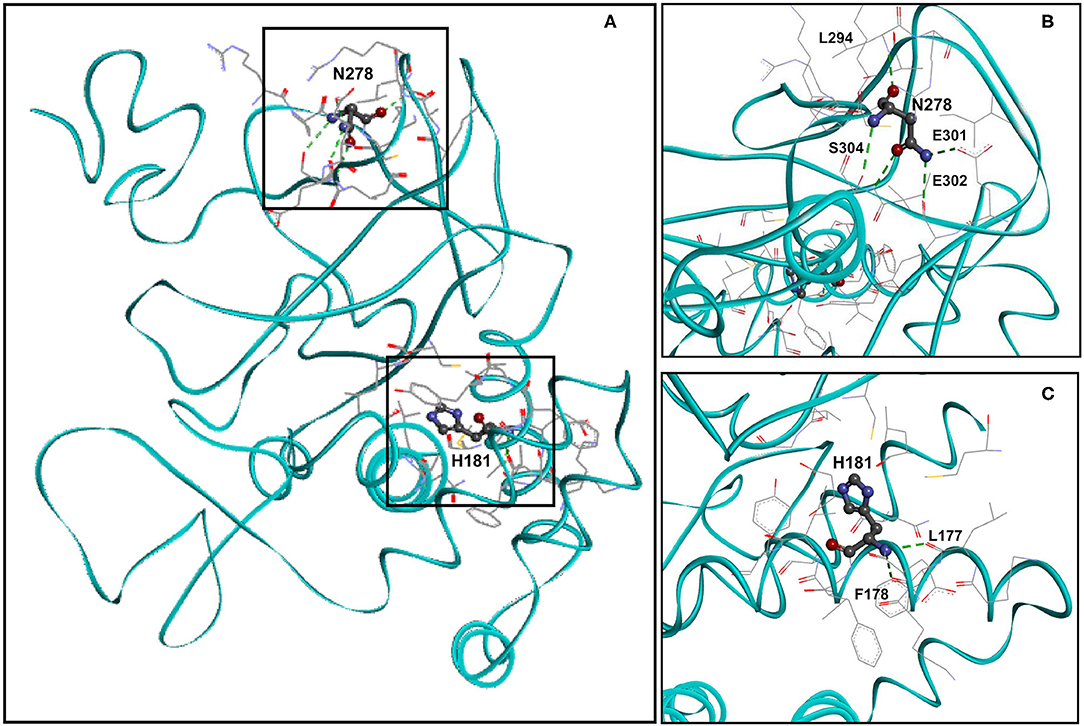
Figure 2. Molecular model of wild-type KMT5B (A). KMT5B is in cyan. Residues H181 and N278 are shown ball and stick by charge. Residues that are within 5Å distance from H181 or from N278 are shown in stick. Hydrogen bonds are presented in dashed green. (B) Close-up of wild-type residue N278 with residues that are within 5Å distance and the residues with which hydrogen bonds are formed. Five hydrogen bonds, presented in dashed green, are formed between N279 and residues L294, E301, E302 and S304. Two hydrogen bonds are formed between N278 and S304. The set of residues (by element, stick) that are within 5Å from N278 are also shown, including: R276, P277, C279, K280, K292, A293, L294, R295, I297, E301, E302, and S304. (C) Close-up of wild-type residue H181 with residues that are within 5Å distance and the residues with which hydrogen bonds are formed. Two hydrogen bonds, presented in dashed green, are formed between H181 and residues L177 and F178. The set of residues (by element, stick) that are within 5Å from H181 are also shown, including: K176, L177, F178, K179, E180, V182, F183, I184, Y185, C230, L265, and G266.
An Additional Hydrogen Bond Is Formed Upon p.H181D Variant
We analyzed the spatial location of the identified H181D (p.His181Asp) variant and the hydrogen bonds it forms (Figure 3A). According to the structural model, while WT H181 forms two hydrogen bonds with residues L177 and F178, the mutated H181D model forms an additional bond with I184. In addition, three of the residues that were within 5Å distance from H181 (L176, L265 and G266) are no longer found in such proximity in the H181D mutant structure.
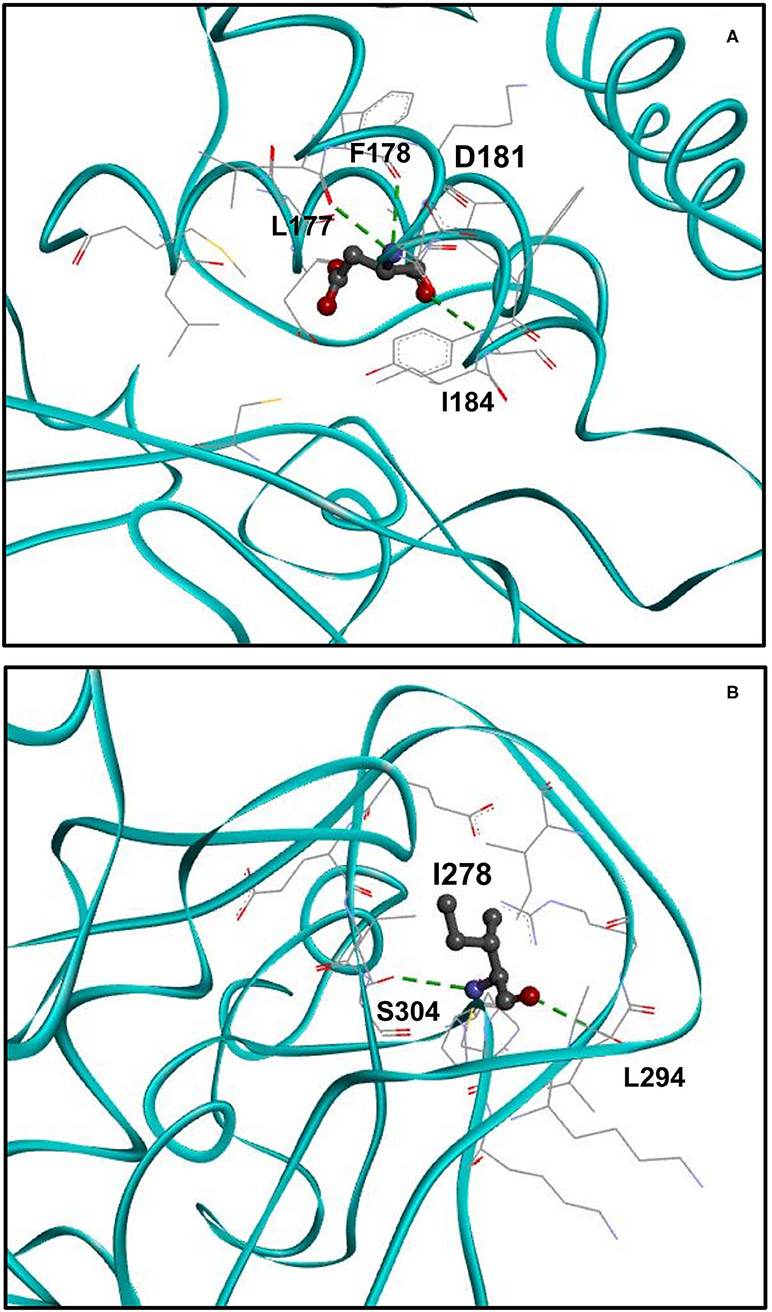
Figure 3. Structure of H181D and N278I mutants. (A) Close up view on the structure of H181D mutant. KMT5B is in cyan. Residue D181 is shown in ball and stick by charge and the residues with which hydrogen bonds are formed, shown in dashed green, are presented in stick. According to the model, hydrogen bonds are formed between D181 and L177, F178 and I184. (B) Close up view on the structure of N278I mutant. Residue I278 is shown in ball and stick by charge and the residues with which hydrogen bonds are formed, shown in dashed green, are presented in stick. Two hydrogen bonds are formed between I278 and S304 and a single hydrogen bond is formed with L294.
Three Hydrogen Bonds Are Lost Upon p.N278I Variant
Finally, we analyzed the spatial location of the identified N278I (p.Asn278Ile) variant and its hydrogen bonds (Figure 3B). While WT N278 forms five hydrogen bonds with residues L294, E301, E302 and two hydrogen bonds with S304, the mutated N278I model forms only two hydrogen bonds. That is, one of the hydrogen bonds with S304 is lost and the hydrogen bonds with E301 and E302 are lost as well. In addition, I303 that was not within 5Å distance from N278 is found to be within the 5Å distance in the N278I mutant structure.
Discussion
The association of disease-causing variants in KMT5B with neurodevelopmental disorders, with or without ASD and/or seizures, has only recently been recognized (4, 6, 7). In this study, we report three individuals with de novo variants in this gene, characterized also by macrocephaly and ophthalmological involvement.
The enzyme KMT5B, as well as its highly homologous counterpart KMT2C, is a lysine methyltransferase known to mainly affect the K20 site on histone protein H4 (H4K20) (13, 14). There is accumulating evidence establishing the involvement of H4K20 methylation in brain development (9).
The majority of the previously-reported variants in KMT5B are protein-truncating variants (PTVs) causing loss of function. These include 6 indel variants, one nonsense variant and a splice-site variant. Two additional patients harbor a genomic deletion encompassing KMT5B. Given these variants found in affected individuals, the pLI scores and the aforementioned murine models (8), the mechanism of disease for this subgroup of patients is haploinsufficiency. In contrast, three previously-reported patients have protein-altering (missense) variants (PAVs). These might result in an alternate mechanism, such as gain-of-function or a dominant-negative effect, however this remains to be elucidated. As presented in Table 1, there are no striking differences between patients with PTVs vs. PAVs with regard to the degree of intellectual disability or the presence of other phenotypic features (autism, seizures, macrocephaly, overgrowth, etc.). Therefore, no clear genotype-phenotype correlations can be safely deduced based on these small numbers of patients.
Using computational 3D modeling of the KMT5B protein, we demonstrate the predicted effect of the two missense variants on hydrogen bonds with nearby residues and on the overall 3D protein structure, further underscoring their pathogenicity.
About 50 histone lysine methyltransferases are recognized in humans. Most of them contain a SET domain as their catalytic domain (15, 16), and along with the post-SET domains are involved in substrate and cofactor binding (17). A multiple sequence alignment of SET domains from different human histone methyltransferases (18) has shown that residue N278, which is found within the SET domain, is evolutionarily conserved and is located in the vicinity of C275 which binds the Zn ion (18).
Hydrogen bonds are one of the major protein structure determinants and their making and breaking profoundly affects the activity of proteins (19, 20). Previous studies have shown that mutation of F281 to A, as well as other mutations along the SET domain, abolishes histone methyltransferase activity (18). Considering the proximity of N278 to F281 and the loss of three hydrogen bonds upon mutation to I, impaired stability may be implied affecting histone methyltransferase activity. As for the H181D mutation, some predictions (e.g. by InterPro and Pfam) suggest it might be considered part of the SET domain as well, so its effect may also be impaired catalytic activity.
While the neurodevelopmental phenotype associated with KMT5B is variable and has yet to be fully characterized, several of its components are becoming evident. Developmental delay and/or ID of varying severity seems to be universal, and was noted in 16/16 (100%) of the patients for whom there is clinical data available in the literature, including the three reported herein. Additional features reported in some individuals include ASD or autistic traits (7/16, 44%; 5/9 of patients with PTVs, 2/5 with PAVs), hypotonia, and seizures (including febrile seizures) (6/16, 37.5%; 4/9 of PTVs, 1/5 with PAVs). Macrocephaly (or relative macrocephaly) is another characteristic (albeit not universal: 7/16, 44%). Brain MRI findings have been noted occasionally, including corpus callosum hypoplasia, hydrocephalus and cerebral ventricular enlargement. Dysmorphic facial features have been described, however these did not seem to be consistent or lead to a recognizable facial phenotype. Nevertheless, several features did recur among some affected individuals, such as ophthalmological involvement (strabismus in 5/16, 31%, ptosis), broad forehead (5/16, 31%), epicanthal folds, high palate, joint hypermobility and long or conical fingers. Finally, overgrowth of varied extent was noted in 5/16 (31%) of patients.
The differential diagnosis of GDD/ID accompanied by macrocephaly is diverse and includes numerous inherited disorders, such as PTEN hamartoma syndromes, Fragile X and Canavan disease, among others (21). Interestingly, macrocephaly (or relative macrocephaly) has been previously described as a common feature of additional neurodevelopmental disorders associated with variants in other genes of the lysine methyltransferase family, such as KMT2E (22). However, the pathomechanistic association between this group of proteins and abnormal brain size has yet to be elucidated. While macrocephaly and/or overgrowth were not consistently described among patients with KMT5B variants, we suggest that this genetic diagnosis should be entertained in undiagnosed patients with ID and macrocephaly or overgrowth.
Finally, due to the small number of patients with point mutations in KMT5B reported to date, and the limited phenotypic information available for most of them, it is still early to determine whether or not genotype-phenotype correlations exist for this entity, and whether there are recognizable differences in terms of disease severity between patients harboring loss-of-function vs. missense variants. Additionally, while most reported patients were male, the overall small number of patients precludes us from drawing conclusions as to possible sex effects on the phenotype. This is particularly interesting given the aforementioned differential effects by sex suggested by behavioral murine studies (8). Future studies detailing clinical and molecular characteristics of larger cohorts of patients with KMT5B variants will shed light on these unanswered questions.
To conclude, our findings expand the current knowledge regarding the phenotype and variant spectrum of KMT5B-associated ID, and suggest that this entity should be considered in the differential diagnosis of ID accompanied by relative macrocephaly and/or overgrowth.
Data Availability Statement
The data sets presented in this article are not readily available, since parental consent was not obtained for sharing the raw data with public databases. Requests to access the databases should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Boards (IRBs) of the Sheba Medical Center and Beilinson Medical Center. Written informed consent to participate in this study was provided by the participant's legal guardian/next of kin.
Author Contributions
AE, LG, and BP-S initiated the study, recruited patients, analyzed the results, wrote the manuscript, and critically reviewed it. GZ, YG, AR-R, AS, OS-C, NP-S, NR-S, EP, MF, and MS obtained clinical data and analyzed it and critically reviewed the manuscript. OB, IB-J, EJ, and LB interpreted the exome sequencing data. VK provided the 3D protein structures and their analyses. All authors critically reviewed the manuscript.
Conflict of Interest
VK was employed by Bioinformatics Consulting.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors wish to thank the patients and their families for their kind assistance. Clinical whole exome sequencing was supported by the Department of Human Genetics, Israeli Ministry of Health (MOH) Pilot Program.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.844845/full#supplementary-material
References
1. Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. (2012) 13:343–57. doi: 10.1038/nrg3173
2. Rowe EM, Xing V, Biggar KK. Lysine methylation: implications in neurodegenerative disease. Brain Res. (2019) 1707:164–71. doi: 10.1016/j.brainres.2018.11.024
3. Jiang Y, Han Y, Petrovski S, Owzar K, Goldstein DB, Allen AS. Incorporating functional information in tests of excess de novo mutational load. Am J Hum Genet. (2015) 97:272–83. doi: 10.1016/j.ajhg.2015.06.013
4. Stessman HA, Xiong B, Coe BP, Wang T, Hoekzema K, Fenckova M, et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat Genet. (2017) 49:515–26. doi: 10.1038/ng.3792
5. Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. (2014) 515:216–21. doi: 10.1038/nature13908
6. Faundes V, Newman WG, Bernardini L, Canham N, Clayton-Smith J, Dallapiccola B, et al. Histone lysine methylases and demethylases in the landscape of human developmental disorders. Am J Hum Genet. (2018) 102:175–87. doi: 10.1016/j.ajhg.2017.11.013
7. Trinh J, Kandaswamy KK, Werber M, Weiss MER, Oprea G, Kishore S, et al. Novel pathogenic variants and multiple molecular diagnoses in neurodevelopmental disorders. J Neurodev. Disord. 11:11. doi: 10.1186/s11689-019-9270-4
8. Wickramasekara RN, Robertson B, Hulen J, Hallgren J, Stessman HAF. Differential effects by sex with Kmt5b loss. Autism Res. (2021) 14:1554–71. doi: 10.1002/aur.2516
9. Wickramasekara RN, Stessman HAF. Histone 4 lysine 20 methylation: a case for neurodevelopmental disease. Biology (Basel). (2019) 8:11. doi: 10.3390/biology8010011
10. Wang ZJ, Rein B, Zhong P, Williams J, Cao Q, Yang F, et al. Autism risk gene KMT5B deficiency in prefrontal cortex induces synaptic dysfunction and social deficits via alterations of DNA repair and gene transcription. Neurophyscopharmacology. (2021) 46:1617–26. doi: 10.1038/s41386-021-01029-y
11. Pode-Shakked B, Barel O, Singer A, Regev M, Poran H, Eliyahu A, et al. A single center experience with publicly funded clinical exome sequencing for neurodevelopmental disorders or multiple congenital anomalies. Sci Rep. (2021) 11:19099. doi: 10.1038/s41598-021-98646-w
12. Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acid Res. (2018) 46:W296–303. doi: 10.1093/nar/gky427
13. Schotta G, Sengupta R, Kubicek S, Malin S, Kauer M, Callén E, et al. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev. (2008) 22:2048–61. doi: 10.1101/gad.476008
14. Hyun K, Jeon J, Park K, Kim J. Writing, erasing and reading histone lysine methylations. Exp Mol Med. (2017) 49:e324. doi: 10.1038/emm.2017.11
15. Min JR, Feng Q, Li Z, Zhang Y, Xu RM. Structure of the catalytic domain of human DOT1L, a Non-SET domain nucleosomal histone methyltransferase. Cell. (2003) 112:711–23. doi: 10.1016/S0092-8674(03)00114-4
16. Lachner M, Jenuwein T. The many faces of histone lysine methylation. Curr Opin Cell Biol. (2002) 14:286–98. doi: 10.1016/S0955-0674(02)00335-6
17. Dillon SC, Zhang X, Trievel RC, Cheng X. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. (2005) 6:227. doi: 10.1186/gb-2005-6-8-227
18. Wu H, Siarheyeva A, Zeng H, Lam R, Dong A, Wu XH, et al. Crystal structures of the human histone H4K20 methyltransferases SUV420H1 and SUV420H2. FEBS Lett. (2013) 587:3859–68. doi: 10.1016/j.febslet.2013.10.020
19. Bowie JU. Membrane protein folding: how important are hydrogen bonds? Curr Opin Struct Biol. (2011) 21:42–9. doi: 10.1016/j.sbi.2010.10.003
20. Pace CN, Fu H, Lee Fryar K, Landua J, Trevino SR, Schell D, et al. Contribution of hydrogen bonds to protein stability. Protein Sci. (2014) 23:652–61. doi: 10.1002/pro.2449
21. Accogli A, Geraldo AF, Piccolo G, Riva A, Scala M, Balagura G, et al. Diagnostic approach to macrocephaly in children. Front Pediatr. (2022) 9:794069. doi: 10.3389/fped.2021.794069
Keywords: KMT5B, intellectual disability, developmental delay, de novo, macrocephaly
Citation: Eliyahu A, Barel O, Greenbaum L, Zaks Hoffer G, Goldberg Y, Raas-Rothschild A, Singer A, Bar-Joseph I, Kunik V, Javasky E, Staretz-Chacham O, Pode-Shakked N, Bazak L, Ruhrman-Shahar N, Pras E, Frydman M, Shohat M and Pode-Shakked B (2022) Refining the Phenotypic Spectrum of KMT5B-Associated Developmental Delay. Front. Pediatr. 10:844845. doi: 10.3389/fped.2022.844845
Received: 28 December 2021; Accepted: 10 February 2022;
Published: 30 March 2022.
Edited by:
Andrea Lynne Gropman, Children's National Hospital, United StatesReviewed by:
Victor Faundes, University of Chile, ChileAndrea Accogli, McGill University Health Centre, Canada
Copyright © 2022 Eliyahu, Barel, Greenbaum, Zaks Hoffer, Goldberg, Raas-Rothschild, Singer, Bar-Joseph, Kunik, Javasky, Staretz-Chacham, Pode-Shakked, Bazak, Ruhrman-Shahar, Pras, Frydman, Shohat and Pode-Shakked. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ben Pode-Shakked, YmVuX3BvZGVAaG90bWFpbC5jb20=