
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 10 March 2022
Sec. Pediatric Gastroenterology, Hepatology and Nutrition
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.842820
This article is part of the Research Topic Hirschsprung Disease: Genetic Susceptibility, Disease Mechanisms and Innovative Management in the Multi-Omics Era View all 5 articles
 Kristy Iskandar1Susan Simanjaya2Taufik Indrawan2
Kristy Iskandar1Susan Simanjaya2Taufik Indrawan2 Alvin Santoso Kalim2
Alvin Santoso Kalim2 Marcellus2
Marcellus2 Didik Setyo Heriyanto3
Didik Setyo Heriyanto3 Gunadi2*
Gunadi2*
Background: Hirschsprung disease (HSCR) is a heterogeneous genetic disease characterized by the absence of ganglion cells in the intestinal tract. The REarranged during Transfection (RET) is the most responsible gene for its pathogenesis. RET’s somatic mosaicisms have been reported for HSCR; however, they are still under-recognized. Therefore, we determined the frequency of somatic mutation of RET rs2435357 in HSCR patients at our institution.
Methods: We performed RET rs2435357 genotyping from 73 HSCR formalin-fixed and paraffin-embedded (FFPE) rectal and 60 non-HSCR controls using the PCR-RFLP method. Subsequently, we compared those frequencies of genotypes for RET rs2435357 with our previous genotyping data from 93 HSCR blood specimens.
Results: The frequencies of genotypes for RET rs2435357 in HSCR paraffin-embedded rectal were CC 0, CT 11 (15%), and TT 62 (85%), whereas their frequencies in HSCR blood samples were CC 4 (4.3%), CT 22 (23.7%), and TT 67 (72%). Those frequencies differences almost reached a significant level (p = 0.06). Moreover, the frequency of RET rs2435357 risk allele (T) was significantly higher in HSCR patients (135/146, 92.5%) than controls (46/120, 38.3%) (p = 3.4 × 10–22), with an odds ratio of 19.74 (95% confidence interval = 9.65–40.41).
Conclusion: Our study suggests somatic mosaicism in HSCR patients. These findings further imply the complexity of the pathogenesis of HSCR. Moreover, our study confirms the RET rs2435357 as a significant genetic risk factor for HSCR patients.
Hirschsprung disease (HSCR) is the leading cause of functional intestinal obstruction in neonates, with 15, 28, and 21 cases per 1,00,000 live births in the European, Asian, and African populations (1, 2). It is caused by the failure of migration, proliferation, and differentiation of neural crest cells during enteric nervous system development (1, 2).
At least 24 genes play a role in the pathogenesis of HSCR, with REarranged during Transfection (RET) as one of the significant genes (1–3). Given the heterogeneity of the genes mentioned above, most genes only have a small effect on the formation of HSCR, which is not more than 20 percent of all patients (1, 2, 4). On the other hand, polymorphism on the intron 1 enhancer gene, RET rs2435357, is found in ∼80% of patients with HSCR (4). This variant is more commonly found with up to 60 percent of patients without a mutation in the coding sequence of RET compared to 14% of patients with a mutation on the coding sequence of RET, such that it is said to be a significant risk factor for male patients with isolated S-HSCR (4). Our previous studies showed that RET rs2435357 variant is a significant risk factor toward the development of the HSCR in Indonesia (5–7).
Several studies suggested the role of somatic mosaicism in HSCR (8, 9). However, the evidence is still limited and controversial (10, 11). Therefore, we aimed to investigate the frequency of somatic mutation of RET rs2435357 in HSCR patients at our institution.
Our samples were the paraffin blocks of rectal tissue from 73 HSCR patients <18 years old and 60 non-HSCR patients <18 years old at our institution. This study was approved by the Medical and Health Research Ethics Committee, Faculty of Medicine, Public Health and Nursing, Universitas Gadjah Mada/Dr. Sardjito Hospital, Yogyakarta, Indonesia (Ref. KE/FK/0855/EC/2017). The research has been performed following the Declaration of Helsinki.
Genomic DNA was extracted from the formalin-fixed and paraffin-embedded (FFPE) rectal of HSCR patients and non-HSCR patients using the QIAmp DNA Mini Kit (QIAGEN, Hilden, Germany). For comparison, we used our previous genotyping data of RET rs2435357 of HSCR patients from blood samples (6). The blood samples and rectal samples were from the same HSCR patients. The HSCR patient samples were from the full-thickness rectal biopsies.
According to our previous study, genotyping of RET rs2435357 variant was done using the PCR-RFLP method using forward primer 5′-gagtgcatggggacagtt-3′ and reverse primer 5′-ggaaactgccaattaggttat-3′ (6). The PCR condition was 95°C for 5 min, 35 cycles (95°C for 1 min, 58°C for 1 min and 72°C for 1 min) and using the PCR Swift Maxi thermal cycler (Esco Micro Pte. Ltd., Singapore). After that, the PCR product was digested using restriction enzyme endonuclease Hin1II (6, 12). The risk T allele will form a restriction site for the abovementioned enzyme to produce a fragment of 156bp and 90bp, whereas the non-risk C allele does not have a restriction location such that it will only produce one fragment of 246bp. Thus, genotype CC will show one band (246bp), CT with three bands (246bp, 156bp, and 90bp), and TT with two bands (156bp and 90bp) on the 3% agarose gel and visualized using ethidium bromide (Figure 1).
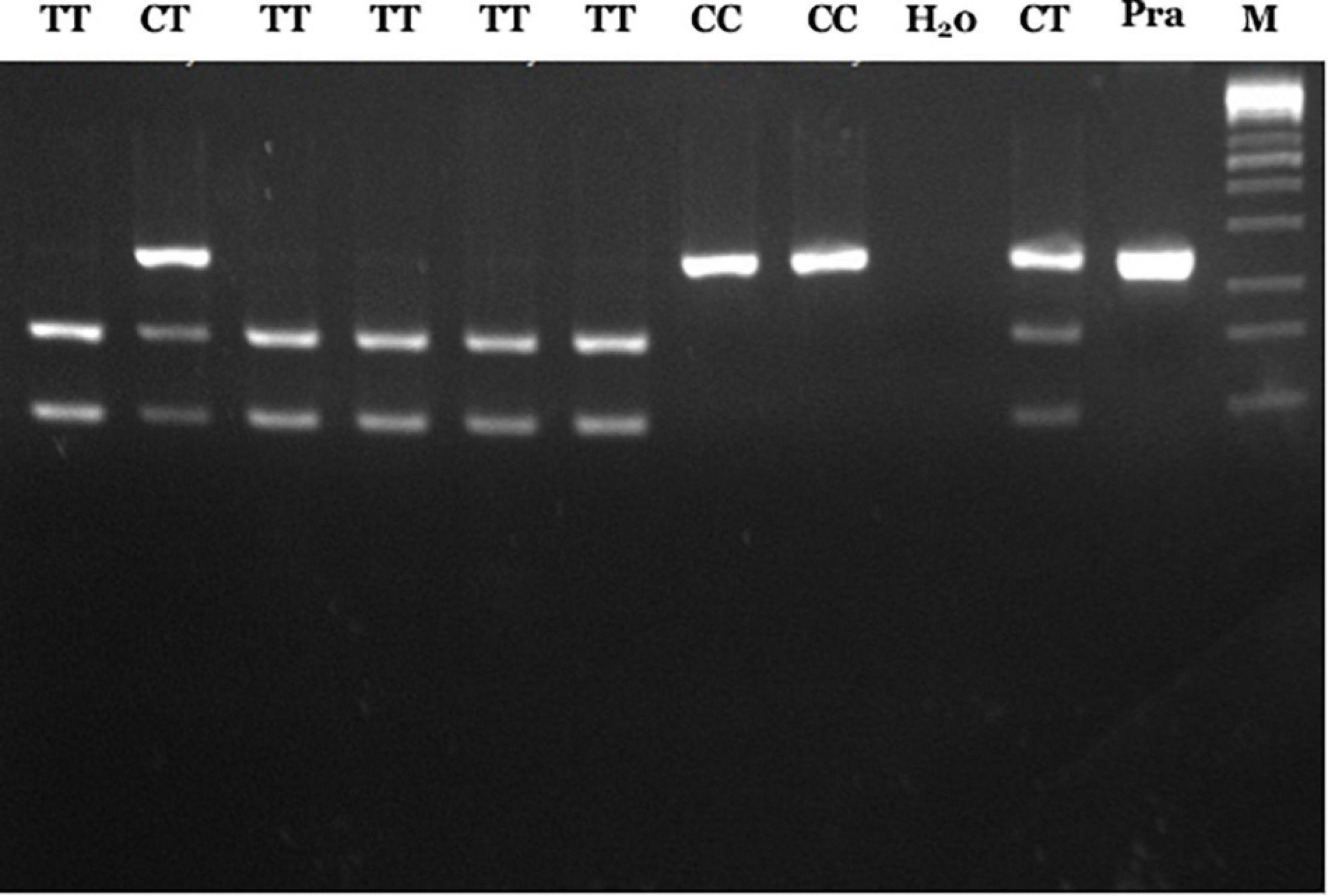
Figure 1. PCR-RFLP results of RET rs2435357 variant. Lane 1, 3–6: TT genotype (156 and 90 bp); lane 2, 10: CT genotype (246, 156, and 90 bp), lane 7–8: CC genotype (246 bp), lane 9: H20, lane 11: pra-digested PCR, and lane M: 100 bp DNA marker.
The association between RET rs2435357 and the risk of HSCR was determined using Chi-square or Fisher Exact test with a significance of p < 0.05.
Firstly, we compared our previous genotype of RET rs2435357 from the blood samples (6) with the rectal tissue. The frequencies of genotypes for RET rs2435357 in HSCR paraffin-embedded rectal tissue were CC 0, CT 11, and TT 62, whereas their frequencies in HSCR blood samples were CC 4, CT 22, and TT 67. Those frequencies differences almost reached a significant level (p = 0.06) (Table 1).

Table 1. Comparison of RET rs2435357 genotype in HSCR patients between rectal and blood samples (6).
Next, we determined the association between RET rs2435357 and the risk of HSCR in our population. The frequency of RET rs2435357 risk allele (T) was significantly higher in HSCR patients (135/146, 92.5%) than controls (46/120, 38.3%) (p = 3.4 × 10–22), with an OR of 19.74 (95% CI = 9.65–40.41) (Table 2). Subsequently, we combined the genotype of RET rs2435357 from rectal and blood samples6 and associated them with the risk of HSCR. Again, the T allele was significantly associated with the HSCR with the OR of 8.11 (95% CI = 5.53–11.88); p = 3.7 × 10–32) (Table 3).
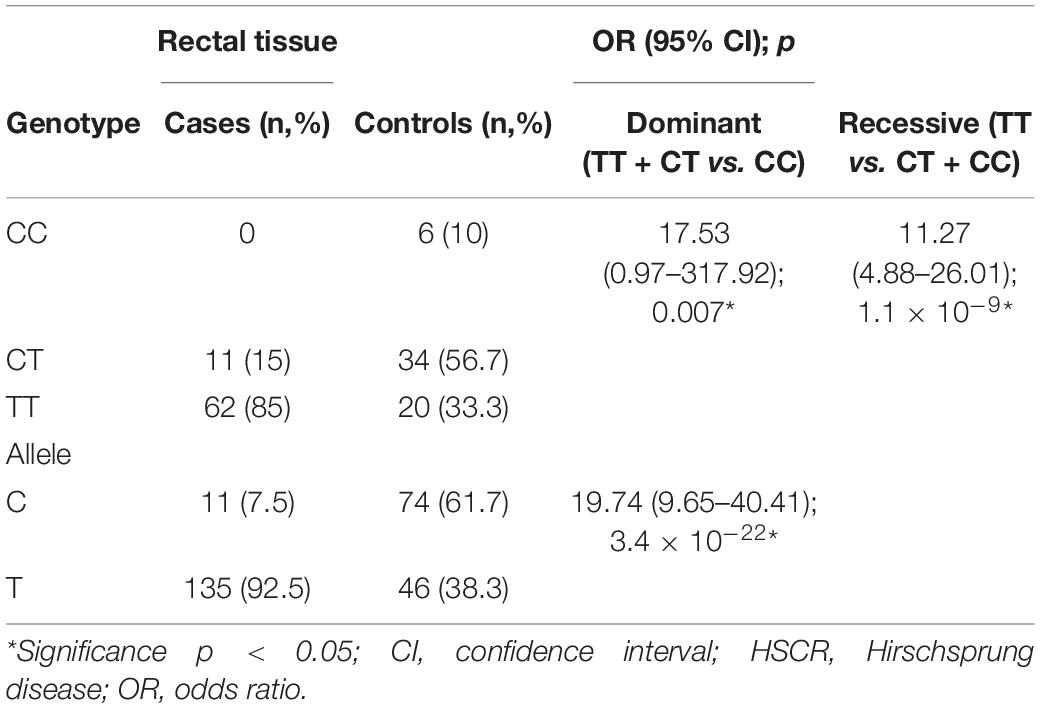
Table 2. Association between RET rs2435357 and risk of HSCR in our study.
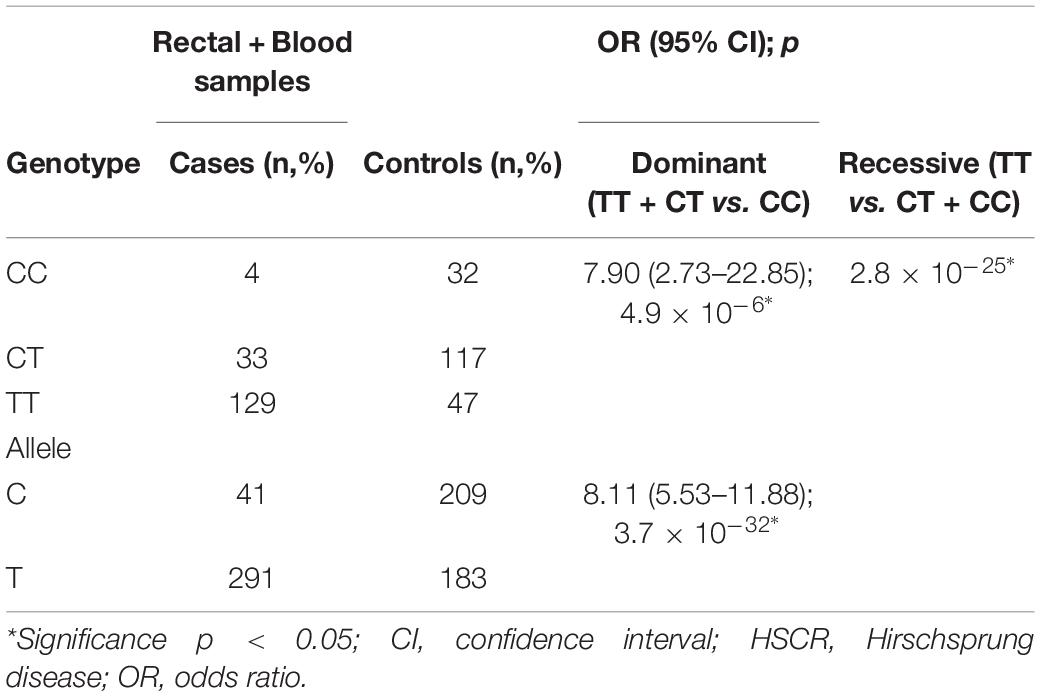
Table 3. Association between RET rs2435357 and risk of HSCR from all samples.
Our study shows that somatic mosaicism might occur in HSCR patients. The role of somatic mosaicism in HSCR is still controversial. While several studies suggested the somatic mosaicism in HSCR (8, 9, 11), a previous report did not (10). Therefore, our findings provided new evidence of somatic mosaicism in HSCR pathogenesis from a different ethnic group, i.e., Javanese, Indonesia. Interestingly, different findings of somatic mosaicism are noted even among the same population, i.e., Chinese (9–11). While two studies from the Chinese population supported somatic mosaicism (9, 11), one report did not (10). As Indonesia consists of more than 375 ethnic groups (5), further studies with a specific another ethnic group in Indonesia are mandatory to clarify the role of somatic mosaicism in the pathogenesis of HSCR in Indonesia. Another difference in our study from previous reports is that we used PCR-RFLP for genotyping of RET rs2435357 in HSCR patients (vs. TaqMan method (10) vs. Sequencing (8). This method has been shown accurate and more affordable than the TaqMan technique to genotype RET rs2435357 in HSCR patients (6).
Most studies of somatic mosaicism focus on cancer (13–15). Somatic mosaicism has been reported in genetic diseases, either Mendelian or complex genetic disorders (8, 9, 11, 14). Somatic mosaicism causes a milder phenotype in Mendelian disorder (14). Whether the somatic mosaicism also results in a milder phenotype in complex genetic disorders such as HSCR is exciting and essential to investigate.
Several pathways have been proposed for the HSCR pathogenesis, including the RET/GFRα1/GDNF, EDNRB/ECE1/EDN3, SOX10/PHOX2B, and SEMA3 (1, 2). However, variants in those pathways attribute to only 20% of all HSCR cases, implying that other mechanisms are supposed to be involved in the pathogenesis of HSCR (16), including somatic mosaicism (9, 11). Although Jiang et al. (9) showed that the somatic mutation of RET has a role in the pathogenesis of HSCR, however, another report did not fully agree with those findings (17). They suggested that to determine the somatic mosaicism in HSCR patients; the study should compare the variants between different tissues representing different germ layers, such as blood and colon tissue (17). Our study compared the frequency of RET rs2435357 variant in HSCR patients from blood and rectal tissue.
Moreover, determining the somatic mosaicism in HSCR is essential to explain the occurrence of HSCR in the absence of inherited or de novo variants during the counseling to the families (18). While the germ-line somatic mutation can be transmitted, the tissue-specific somatic mutation is not (18). A current study failed to identify somatic mosaicism in a small number of HSCR patients. They suggested that it is challenging to find the somatic variants involved in HSCR because these mutations will lead to a selective disadvantage for the affected cell (18).
This study focused on the RET rs2435357 variant since this variant has been a vital genetic risk factor for HSCR across populations, including Indonesia (4–7, 19–21). Our current study also supports the RET rs2435357 as a significant risk factor for HSCR (Tables 2, 3). RET rs2435357 reduces the binding of the critical transcription factor (TF) SOX10 necessary for ganglionosis during the enteric nervous system development (4). These mechanisms are in harmonizing with two other enhancers in RET: one binding TF GATA2 and the other binding TF RARB (22). In addition, recent meta-analysis studies showed that besides RET rs2435357, other variants in RET also increased HSCR risk, including rs1800858, rs1800861, and rs10900297 (23, 24). Further study is necessary to investigate the somatic mosaicism of those three RET variants to confirm our findings.
Notably, we extracted the DNA from the FFPE rectal samples. It might result in non-reproducible sequence artifacts (10). In addition, we genotyped the RET rs2435357 variant from the rectal samples only. Further study is necessary to use the fresh tissue and compare the somatic mosaicism status between aganglionic, ganglionic, and transitional colon samples.
Our study suggests somatic mosaicism in HSCR patients. These findings further imply the complexity of the pathogenesis of HSCR. Moreover, our study confirms the RET rs2435357 as a significant genetic risk factor for HSCR patients.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the Medical and Health Research Ethics Committee, Faculty of Medicine, Public Health and Nursing, Universitas Gadjah Mada/Dr. Sardjito Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
KI and Gunadi conceived the study. KI, Gunadi, SS, and Marcellus drafted the manuscript. SS, TI, and Marcellus collected the data. Gunadi analyzed the data. KI, TI, DH, and Gunadi facilitated all project-related tasks. All authors read and approved the final manuscript.
This study was funded by the Faculty of Medicine, Public Health and Nursing, Universitas Gadjah Mada.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We extend our thanks to all those who provided excellent technical support and assistance during the study.
1. Alves M, Sribudiani Y, Brouwer R, Amiel J, Antiñolo G, Borrego S, et al. Contribution of rare and common variants determine complex diseases—Hirschsprung disease as a model. Dev Biol. (2013) 382:320–9. doi: 10.1016/j.ydbio.2013.05.019
2. Tilghman J, Ling A, Turner T, Sosa M, Krumm N, Chatterjee S, et al. Molecular genetic anatomy and risk Profile of Hirschsprung’s disease. N Engl J Med. (2019) 380:1421–32. doi: 10.1056/NEJMoa1706594
3. Amiel J, Sproat-Emison E, Garcia-Barcelo M, Lantieri F, Burzynski G, Borrego S, et al. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet. (2008) 45:1–14. doi: 10.1136/jmg.2007.053959
4. Emison E, Garcia-Barcelo M, Grice E, Lantieri F, Amiel J, Burzynski G, et al. Differential contributions of rare and common, coding and noncoding ret mutations to multifactorial Hirschsprung disease liability. Am J Hum Genet. (2010) 87:60–74. doi: 10.1016/j.ajhg.2010.06.007
5. Gunadi Kapoor A, Ling A, Rochadi Makhmudi A, Herini E, et al. Effects of RET and NRG1 polymorphisms in Indonesian patients with Hirschsprung disease. J Pediatr Surg. (2014) 49:1614–8. doi: 10.1016/j.jpedsurg.2014.04.011
6. Gunadi Dwihantoro A, Iskandar K, Makhmudi A, Rochadi Accuracy of polymerase chain reaction-restriction fragment length polymorphism for RET rs2435357 genotyping as Hirschsprung risk. J Surg Res. (2016) 203:91–4. doi: 10.1016/j.jss.2016.02.039
7. Gunadi Iskandar K, Makhmudi A, Kapoor A. Combined genetic effects of RET and NRG1 susceptibility variants on multifactorial Hirschsprung disease in Indonesia. J Surg Res. (2019) 233:96–9. doi: 10.1016/j.jss.2018.07.067
8. Moore S, Zaahl M. Tissue specific somatic mutations and aganglionosis in Hirschsprung’s disease. J Pediatr Surg. (2014) 49:258–61. doi: 10.1016/j.jpedsurg.2013.11.035
9. Jiang Q, Liu F, Miao C, Li Q, Zhang Z, Xiao P, et al. RET somatic mutations are underrecognized in Hirschsprung disease. Genet Med. (2018) 20:770–7. doi: 10.1038/gim.2017.178
10. Zhang Z, Jiang Q, Li Q, Cheng W, Qiao G, Xiao P, et al. Genotyping analysis of 3 RET polymorphisms demonstrates low somatic mutation rate in Chinese Hirschsprung disease patients. Int J Clin Exp Pathol. (2015) 8:5528–34.
11. Jiang Q, Wang Y, Li Q, Zhang Z, Xiao P, Wang H, et al. Sequence characterization of RET in 117 Chinese Hirschsprung disease families identifies a large burden of de novo and parental mosaic mutations. Orphanet J Rare Dis. (2019) 14:237. doi: 10.1186/s13023-019-1194-2
12. Borun P, Jerzy S, Ziemnicka K, Kubaszewski L, Lipinski D, Plawski A. Absence of the RET+3:T allele in the MTC patients. Hered Cancer Clin Pract. (2012) 10:14. doi: 10.1186/1897-4287-10-14
13. Fernández L, Torres M, Real F. Somatic mosaicism: on the road to cancer. Nat Rev Cancer. (2016) 16:43–55. doi: 10.1038/nrc.2015.1
14. Cohen A, Wilson S, Trinh J, Ye X. Detecting somatic mosaicism: considerations and clinical implications. Clin Genet. (2015) 87:554–62. doi: 10.1111/cge.12502
15. Biesecker L, Spinner N. A genomic view of mosaicism and human disease. Nat Rev Genet. (2013) 14:307–20. doi: 10.1038/nrg3424
16. Luzón-Toro B, Villalba-Benito L, Torroglosa A, Fernández R, Antiñolo G, Borrego S. What is new about the genetic background of Hirschsprung disease? Clin Genet. (2020) 97:114–24. doi: 10.1111/cge.13615
17. Brosens E, MacKenzie K, Alves M, Hofstra R. Do RET somatic mutations play a role in Hirschsprung disease? Genet Med. (2018) 20:1477–8. doi: 10.1038/gim.2018.6
18. MacKenzie KC, Garritsen R, Chauhan RK, Sribudiani Y, de Graaf BM, Rugenbrink T. The somatic mutation paradigm in congenital malformations: Hirschsprung disease as a model. Int J Mol Sci. (2021) 22:12354. doi: 10.3390/ijms222212354
19. Jiang Q, Wang Y, Gao Y, Wang H, Zhang Z, Li Q, et al. RET compound inheritance in Chinese patients with Hirschsprung disease: lack of penetrance from insufficient gene dysfunction. Hum Genet. (2021) 140:813–25. doi: 10.1007/s00439-020-02247-y
20. Virtanen V, Salo P, Cao J, Löf-Granström A, Milani L, Metspalu A, et al. Noncoding RET variants explain the strong association with Hirschsprung disease in patients without rare coding sequence variant. Eur J Med Genet. (2019) 62:229–34. doi: 10.1016/j.ejmg.2018.07.019
21. Kapoor A, Jiang Q, Chatterjee S, Chakraborty P, Sosa M, Berrios C, et al. Population variation in total genetic risk of Hirschsprung disease from common RET. SEMA3 and NRG1 susceptibility polymorphisms. Hum Mol Genet. (2015) 24:2997–3003. doi: 10.1093/hmg/ddv051
22. Chatterjee S, Kapoor A, Akiyama J, Auer D, Lee D, Gabriel S, et al. Enhancer variants synergistically drive dysfunction of a gene regulatory network in Hirschsprung disease. Cell. (2016) 167:355.68.e10. doi: 10.1016/j.cell.2016.09.005
23. Amooee A, Lookzadeh MH, Mirjalili SR, Miresmaeili SM, Aghili K, Zare-Shehneh M. Association of rs2435357 and rs1800858 polymorphisms in RET proto-oncogene with Hirschsprung disease: systematic review and meta-analysis. Arq Bras Cir Dig. (2019) 32:e1448. doi: 10.1590/0102-672020190001e1448
Keywords: Hirschsprung disease, RET rs2435357 variant, pathogenesis, somatic mosaicism, specific tissue expression
Citation: Iskandar K, Simanjaya S, Indrawan T, Kalim AS, Marcellus, Heriyanto DS and Gunadi (2022) Is There Any Mosaicism in REarranged During Transfection Variant in Hirschsprung Disease’s Patients? Front. Pediatr. 10:842820. doi: 10.3389/fped.2022.842820
Received: 24 December 2021; Accepted: 21 February 2022;
Published: 10 March 2022.
Edited by:
Consolato M. Sergi, Children’s Hospital of Eastern Ontario (CHEO), CanadaReviewed by:
Roger Leng, University of Alberta, CanadaCopyright © 2022 Iskandar, Simanjaya, Indrawan, Kalim, Marcellus, Heriyanto and Gunadi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gunadi, ZHJndW5hZGlAdWdtLmFjLmlk
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.