 JinFang Zhang
JinFang Zhang LingJi Zeng2
LingJi Zeng2 YuLian Wang
YuLian Wang- 1Department of Paediatric Hematology, Guangdong Provincial People's Hospital, Guangdong Academy of Medical Sciences, Guangzhou, China
- 2Department of Hematology, Guangdong Provincial People's Hospital, Guangdong Academy of Medical Sciences, Guangzhou, China
Objective: To investigate the correlation between gene mutations and glucocorticoid resistance in pediatric acute lymphoblastic leukemia (ALL).
Methods: A total of 71 children with ALL admitted to our center between September 2019 and September 2021 were enrolled. DNA obtained from bone marrow or peripheral blood samples at initial diagnosis was used for genetic testing via whole exome sequencing. Meanwhile, patient clinical information was collected. Subsequently, the correlations of gene mutations with clinical features and glucocorticoid resistance were analyzed.
Results: Of the 71 children enrolled, 61 (85.9%) had B-cell ALL (B-ALL) and 10 (14.1%) had T-cell ALL (T-ALL). The five genes with the highest mutation frequency in B-ALL were TTN (24.4%), FLT3 (14.6%), TP53 (14.6%), MUC16 (9.8%), and EPPK1 (9.8%). In contrast, those with the highest frequency in T-ALL were NOTCH1 (54.5%), FBXW7 (27.3%), TTN (27.3%), MUC16 (27.3%), and PHF6 (18.2%). Upon statistical analysis, TTN and NOTCH1 mutations were found to be associated with prednisone resistance. Further, TTN and MUC16 mutations were associated with a lower age at diagnosis, and NOTCH1 mutations were associated with T-ALL in female patients. Leukocyte counts and LDH levels did not differ based on the presence of any common gene mutation, and no association between these gene mutations and overall survival was observed.
Conclusions: Our study is the first to demonstrate the association between TTN mutation and glucocorticoid resistance in ALL. Our findings could guide strategies for overcoming drug resistance and aid in the development of drug targets.
Introduction
Pediatric acute lymphoblastic leukemia (ALL) is the most common malignancy in children and adolescents, accounting for ~25% of tumors in children aged ≤15 years (1). Due to the development of combination chemotherapy, targeted therapy, cell therapy, and hematopoietic stem cell transplantation, the prognosis of pediatric ALL has improved considerably. The overall survival (OS) for pediatric ALL in individuals with standard risk is now more than 80%, and St. Jude Children's research hospital reported a 5-year OS of 93.5% in this group (2).
Nevertheless, there remain cases of ALL that cannot be cured. In these cases, refractory disease or relapse is mainly a result of leukemia cell resistance (3). Currently, the diagnostic stratification of ALL is based on a combination of clinical information and morphology, immunology, cytology, and molecular biology (MICM) and can be used for comprehensive prognostication. Meanwhile, early treatment response is also an independent predictor of prognosis in pediatric ALL. Poor treatment response, especially for patients with glucocorticoid resistance, often predicts poor prognosis (4). Several studies have found that certain molecular abnormalities are associated with a poor prognosis and drug resistance. For instance, IKZF1 deletions are associated with tyrosine kinase inhibitor resistance in Philadelphia chromosome-positive (Ph+) leukemia (5). Furthermore, CDKN2 deletions are associated with chemotherapy resistance in ALL (6). As for the mechanism of glucocorticoid resistance, more and more molecular abnormalities have been discovered, for example, CREBBP and NT5C1 mutations have been reported to be associated with glucocorticoid resistance (7), however, there are still far more unknown. Therefore, a more detailed molecular understanding can help us screen high-risk patients at initial diagnosis, allowing early intervention and improvements in the cure rate among pediatric patients, and molecular information has become an important part of diagnosis and risk stratification. In this study, MICM typing and gene mutations were analyzed in ALL patients admitted to our center in order to identify the gene mutations associated with glucocorticoid resistance in ALL, and guild advancements in molecular diagnosis/risk stratification and drug target screening for ALL.
Methods
Clinical Data
This study was reviewed by the ethics committee of Guangdong Provincial People's Hospital and Guangdong Academy of Medical Sciences. Samples were collected after obtaining informed consent from the patients' guardians. A total of 71 children with ALL admitted to our center between September 2019 and September 2021 were enrolled. Bone marrow or peripheral blood samples collected at initial diagnosis were used for genetic testing via whole exome sequencing (WES). Meanwhile, fluorescence in situ hybridization (FISH), flow cytometry, karyotyping, RT-PCR analysis, routine laboratory tests, and physical examination were also performed. Treatment was started immediately upon diagnosis. The treatment schedule followed the SCCLG-ALL-2016 protocol1. This protocol was initiated with a prednisone test at a dose of 60 mg/m2·d for 7 consecutive days or >200 mg/m2 week. Prednisone sensitivity was determined according to the SCCLG-ALL-2016 protocol; that is, a primary naive lymphocyte count of <1 × 109/L in peripheral blood at the end of the prednisone test was considered an indicator of sensitivity, whereas a value >1 × 109/L was considered an indicator of resistance. Cerebrospinal fluid (CSF) was examined using cell smears and flow cytometric analysis. The diagnosis of central nervous system (CNS) leukemia was based on the SCCLG-ALL-2016 protocol.
DNA Sequencing
Before treatment, 2 mL bone marrow or peripheral venous blood samples were obtained from the ALL patients. EDTA was added as an anticoagulant, and DNA was extracted from the collected samples. Whole exome gene sequencing was performed to screen gene mutations via the ALL DNA whole exome sequencing gene chip (Illumina) on an Illumina sequencer (Illumina Nextseq500, US). The STRING online software (https:www.string-db.org) was used to conduct signaling pathway analysis for all identified gene mutations.
Follow-Up
Follow-up was conducted via hospital visits or telephone discussions. The follow-up continued until September 30, 2021. Overall Survival was defined as the duration between diagnosis and death or the final follow-up.
Statistical Analysis
We used SPSS 13.0 software for statistical analysis. Comparisons of categorical variables were performed using Pearson's Chi-squared test or Fisher's exact test. OS was determined using Kaplan–Meier analysis. P < 0.05 was considered statistically significant.
Results
Clinical Features
A total of 71 children (age, 1–14 years) with ALL confirmed using bone marrow smears underwent MICM subtyping. Ten patients (14.1%) had T-cell ALL (T-ALL) and 61 (85.9%) had B-cell ALL (B-ALL). There were 43 boys and 28 girls, with a male to female ratio of 1.5:1. The ranges of white blood cell (WBC) counts and lactate dehydrogenase (LDH) levels were 0.98–413 × 109/L and 101–16863 U/L, respectively. Karyotype analysis revealed a normal karyotype in 24 patients (33.8%) and abnormal karyotype in 36 patients (50.7%); however, no chromosomal information was obtained for 11 patients (15.4%). Fusion gene detection based on FISH and RT-PCR revealed no abnormalities in 47 patients (66.2%). However, 8 patients (11.2%) were TEL-AML1+, 5 (7.0%) were BCR-ABL+, 5 (7.0%) were E2A-PBX1+, 2 (2.8%) were KMT2A +, 2 (2.8%) were HOX11+, 1 (1.4%) was TEL-ABL+, and 1 (1.4%) had a TEL deletion. Seven patients (9.8%) were diagnosed with CNS leukemia (e.g., CNS2 and CNS3), including 3 with T-ALL, 4 with B-ALL, 1 with BCR-ABL+ disease, and 1 with TEL-ABL+ disease. All cases of relapse (7, 9.8%) involved B-cell bone marrow relapse; these cases included 1 case of E2A-PBX1+ disease, 1 of HOX11+ disease, 3 of BCR-ABL+ disease, and 1 of TEL-AML1+ disease (Table 1).
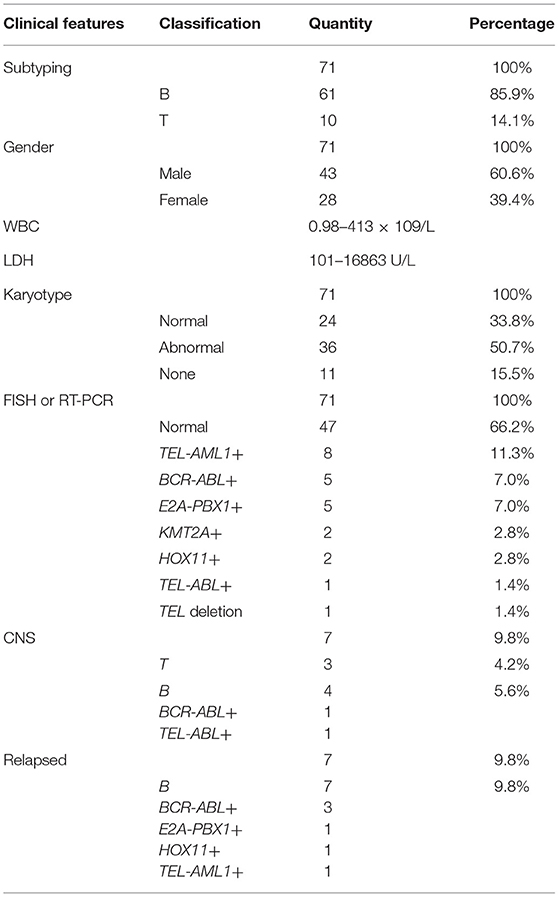
Table 1. Clinical feature of the 71 patients.
Gene Mutation and Signaling Pathway Analysis
Gene mutation analysis revealed 10 genes with a high mutation frequency in B-ALL: TTN (24.4%), FLT3 (14.6%), TP53 (14.6%), MUC16 (9.8%), EPPK1 (9.8%), NOTCH1 (9.8%), CUX1 (9.8%), BRCA2 (9.8%), KMT2C (7.3%), and CSMD1 (7.3%). In T-ALL, the genes with a high mutation frequency were NOTCH1 (54.5%), FBXW7 (27.3%), TTN (27.3%), MUC16 (27.3%), PHF6 (18.2%), KMT2D (18.2%), EPPK1 (18.2%), FLT3 (18.2%), IL7R (18.2%), and JAK3 (7.3%). Of the 7 children with CNS leukemia, 1 was JAK2+, 1 was FLT3+, and 1 was MUC16+ (Figure 1).
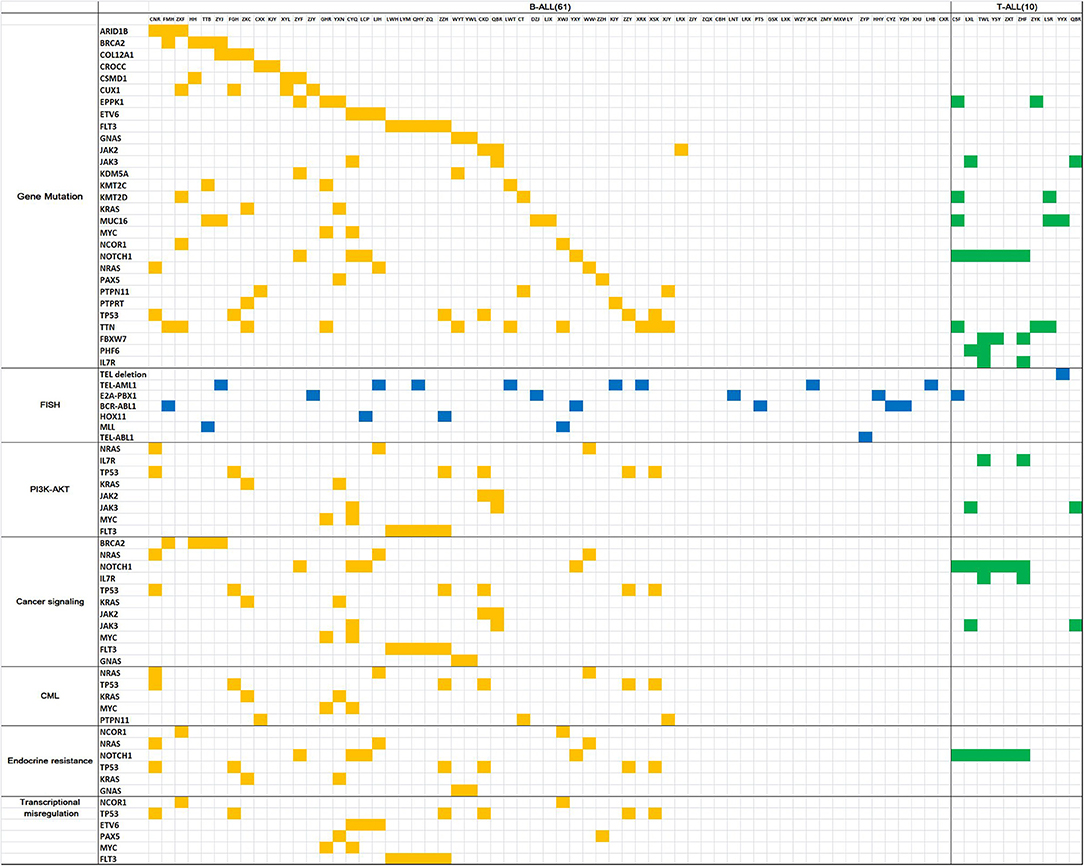
Figure 1. Gene mutation map for acute lymphoblastic leukemia. The horizontal columns represent the patient number, the vertical columns represent the gene name, the yellow squares represent positive gene mutation in B-ALL, the green squares represent positive gene mutationin T-ALL, the blue squares represent positive FISH results.
STRING analysis showed that the primary signaling pathways involved were cancer signaling (BRCA2, NRAS, NOTCH1, IL7R, TP53, KRAS, JAK2, JAK3, MYC, FLT3, and GNAS), cancer MIS transcription signaling (NCOR1, TP53, ETV6, PAX5, MYC, and FLT3), PI3K-Akt signaling (NRAS, IL7R, TP53, KRAS, JAK2, JAK3, MYC, and FLT3), endocrine resistance signaling (NCOR1, NRAS, NOTCH1, TP53, KRAS, and GNAS), and chronic myeloid leukemia signaling (NRAS, TP53, KRAS, MYC, and PTPN11) (Figure 1).
Correlation of Gene Mutations With Prednisone Sensitivity
There were 13 children with TTN+ disease, accounting for 18.3% of all patients. Among them, 3 had T-ALL and 10 had B-ALL. One was E2A-PBX1+, 1 was KMT2A+, 2 were TEL-AML1+, and 1 was BCR-ABL+. The prednisone test revealed sensitivity in 6 children and resistance in 7 children (Table 2). Among the TTN- children, the prednisone test revealed sensitivity in 48 children and resistance in 10 children. The difference in prednisone sensitivity between TTN+ and TTN- children was analyzed using the Chi-square test, and a P-value <0.05 was obtained. Therefore, TTN mutations appeared to be associated with prednisone resistance (Table 4). There were 10 children with NOTCH1+ disease, accounting for 14.08% of all patients. Of them, 6 had T-ALL and 4 had B-ALL. Moreover, 1 was E2A-PBX1+ and 1 was BCR-ABL+. The prednisone test revealed resistance in 6 children and sensitivity in the other 4 (Table 3). Among NOTCH1- children, the prednisone test revealed sensitivity in 50 children and resistance in 11 children. The difference in prednisone sensitivity between NOTCH1+ and NOTCH1- children was analyzed using the Chi-square test, and a P-value <0.05 was obtained. Therefore, NOTCH1 mutations appeared to be associated with prednisone resistance (Table 4).
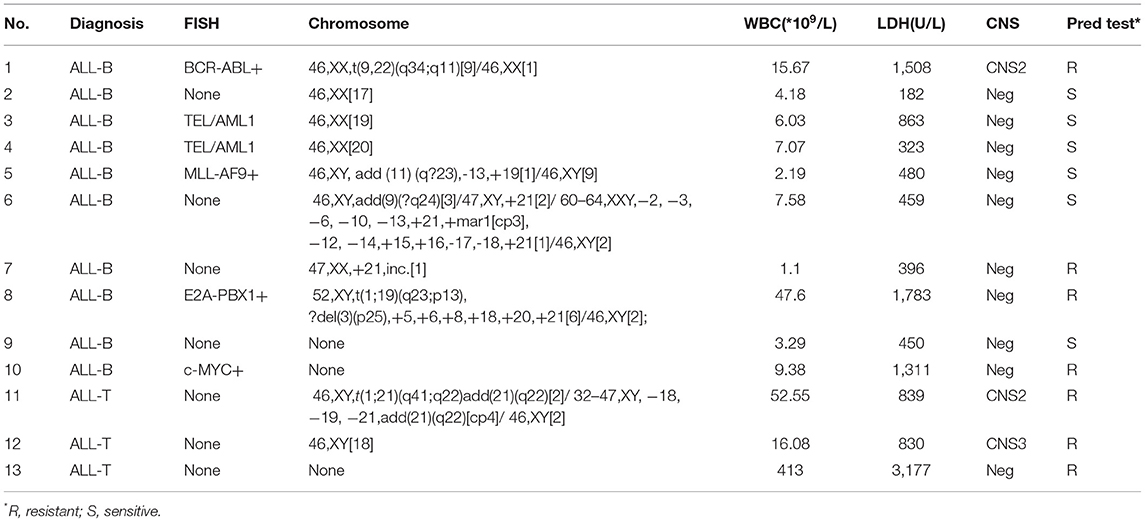
Table 2. Clinical information from patients with TTN+ mutations.

Table 3. Clinical information from patients with NOTCH1+ mutations.
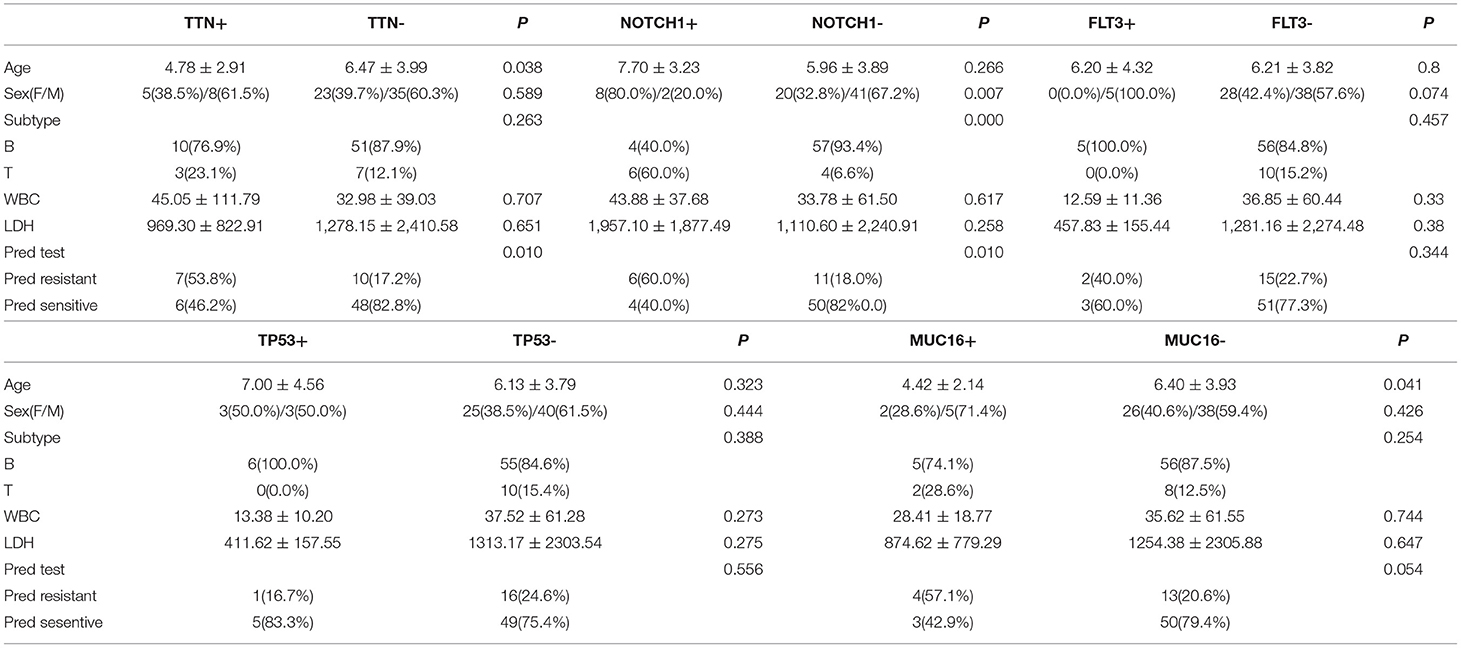
Table 4. Clinical features associated with key gene mutations in pediatric acute lymphoblastic leukemia.
There were 5 children with FLT3+ disease, accounting for 7.04% of all patients; all of them had B-ALL, and one of them was TEL-AML1+. FLT3 mutations did not appear to be associated with prednisone resistance (Chi-square test, P-value >0.05) (Table 4).
There were 6 children with TP53+ disease, accounting for 8.4% of all patients, and all of them had B-ALL without any special fusion genotype. TP53 mutations did not appear to be associated with prednisone resistance (Chi-square test, P-value >0.05) (Table 4).
There were 7 children with MUC16+ disease, accounting for 9.8% of all patients. Of these patients, 2 had T-ALL and 5 had B-ALL. Moreover, 2 were E2A-PBX1+, 1 was KMT2A +, and 1 was TEL-AML1+. MUC16 mutations did not appear to be associated with prednisone resistance (Chi-square test, P-value >0.05) (Table 4).
The relationship of EPPK1, FBXW7, and PHF6 mutations with drug resistance could not be analyzed owing to the small number of cases.
Correlation of Gene Mutations With Clinical Features and Survival
There were no statistically significant differences in leukocyte counts and LDH levels between patients with and without mutations in the following genes (Fisher's exact test, P > 0.05): TTN, NOTCH1, FLT3, TP53, and MUC16. In terms of age, the TTN+ group was younger than the TTN- group (Fisher's exact test, P < 0.05), and the MUC16+ group was younger than the MUC16- group (Fisher's exact test, P < 0.05). In terms of sex, there was a significantly higher proportion of female patients in the NOTCH1+ group than in the NOTCH1- group (Chi-square test, P < 0.05). In terms of disease type, the NOTCH1+ group had a significantly higher proportion of patients with T-ALL than did the NOTCH1- group (Chi-square test, P < 0.05) (Table 4).
MUC16, TP53 and NOTCH1 mutations showed no association with OS (Kaplan–Meier analysis, P > 0.05) (Figure 2). However, this may be a result of the short follow-up and the small number of cases. Groups of TTN and FLT3 mutations could not be analyzed statistically owing to data limitations (Patients with TTN+ or FLT3+ were all alive at the final follow-up date).
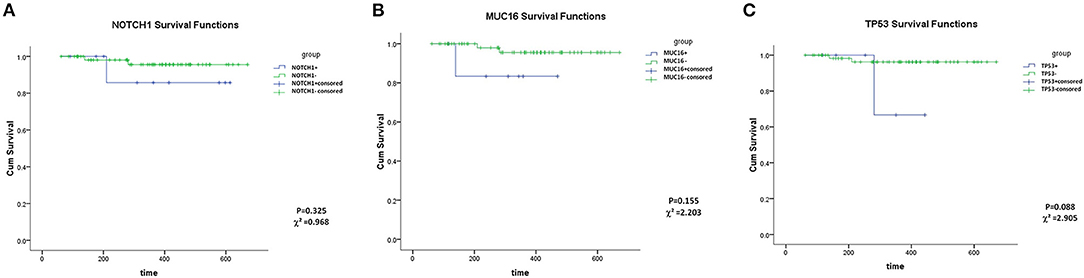
Figure 2. Overall survival analysis of NOTCH1 (A), MUC16 (B), TP53 (C) mutations.
Discussion
Extraordinary advances have been made in the treatment of pediatric ALL, and prognosis has also improved significantly over the last few years. Nevertheless, more than 10% of cases of relapsed or refractory ALL cannot be cured (8). The cost of treatment has significantly increased, creating a burden for families and society at large. Given that the prognosis of pediatric ALL is associated with factors such as early treatment response, karyotype, fusion genes, gene mutations, and minimal residual disease (MRD), molecular diagnosis and risk stratification are important for the early identification of high-risk refractory ALL and the selection of appropriate therapy. Among the clinical features of ALL, an onset age of 1–10 years and a WBC count <50 × 109/L are associated with a good prognosis. In contrast, an onset age of <1 year or >10 years and a WBC count >50 × 109/L are associated with a poor prognosis. With respect to disease subtype, T-ALL is associated with a slightly worse prognosis than B-ALL (9). In terms of karyotype, it has been proven that hyperdiploidy as well as t(12;21) (p13;q22) (ETV6-RUNX1) are associated with a good prognosis, whereas hypodiploidy, KMT2A rearrangements, and Ph-like, IKZF1, and CRLF2 rearrangements are associated with a poor prognosis (10).
Early treatment response is an independent predictor of prognosis in pediatric ALL. The amount of MRD and prednisone sensitivity during induction therapy are important indicators of an early treatment response (11). Studies have shown that the risk of relapse and death is 3–5 times higher in children with MRD >0.01% at the end of induction therapy than in those with MRD <0.01% (12). Prednisone resistance is an independent prognostic factor for the risk-stratified diagnosis and treatment of ALL. Children with leukemia that is resistant to glucocorticoids are considered high risk for treatment failure (13). Glucocorticoids are effective treatment for ALL by leading to direct lymphocyte apoptosis (14). Studies have shown that glucocorticoids mainly induce apoptosis through NR3C1 receptors on the surface of lymphocytes. Mutations in NR3C1 as well as its co-stimulatory molecule CREBBP can lead to the transcriptional dysregulation of genes involved in glucocorticoid action, causing drug resistance (15). The increased expression of some anti-apoptotic molecules, including BCL2, BCL-xL, and MCL1, can also lead to decreased glucocorticoid sensitivity (16). Additionally, abnormalities in the bone marrow microenvironment are also associated with drug resistance. IL7 in the bone marrow microenvironment is believed to induce glucocorticoid resistance in T-ALL cells (17). In terms of signaling pathways, RAS/MEK/ERK, IL7R/JAK/STAT, and PI3K/AKT signaling have been found to be associated with glucocorticoid resistance. Currently, inhibitors targeting these signaling pathways, i.e., MEK inhibitors, AKT inhibitors, and JAK inhibitors, are being tested in clinical trials (18). Nevertheless, the mechanism underlying glucocorticoid resistance remains to be fully elucidated. Therefore, more clinical data as well as basic research are required to explore the mechanisms of drug resistance in ALL.
In this study, we analyzed clinical data, including clinical features, fusion genes, and gene mutation data, obtained from pediatric ALL patients treated at our center over the past 2 years. In terms of subtype distribution as well as sex, our findings were consistent with international and domestic data (19). Using FISH and RT-PCR results, we observed that BCR-ABL fusion occurred in 7% of our patients, which was slightly higher than the rate reported previously (3–5%). TEL-AML1fusion was observed in 11.2% of cases, which was lower than the previously reported rate of 25% (20). These differences may be related to the small number of cases in our study. [HOX11t(10, 14)] (20% in T-ALL), E2A-PBX1 (7%), KMT2A (2.8%) and TEL deletions (1.8%) rates were similar to those reported previously (21–24). All cases of CNS involvement were detected at initial diagnosis or during induction therapy. The incidence of CNS leukemia in our study was 9.8%, higher than the rates of 3–8% reported in previous studies from China and the rest of the world (25, 26). This difference could be related to the small sample size in our study and inadequate sedation during lumbar puncture.
Gene mutation analysis revealed 10 genes with a high gene mutation frequency in T-ALL: NOTCH1 (54.5%), FBXW7 (27.3%), TTN (27.3%), MUC16 (27.3%), PHF6 (18.2%), KMT2D (18.2%), EPPK1 (18.2%), FLT3 (18.2%), IL7R (18.2%), and JAK3 (7.3%).Of these genes„ the mutation frequency for FLT3 mutations were slightly more frequent than previously reported (27–29), MUC16, which showed a high mutation frequency in our study, was less common in previous reports. Moreover, to our knowledge, our study is the first to detect EPPK1 and TTN mutations in T-ALL. The top 10 genes with a high mutation frequency in B-ALL were TTN (24.4%), FLT3 (14.6%), TP53 (14.6%), MUC16 (9.8%), EPPK1 (9.8%), NOTCH1 (9.8%), CUX1 (9.8%), BRCA2 (9.8%), KMT2C (7.3%), and CSMD1 (7.3%). These findings were different from those of previous studies from outside China, which most commonly reported PAX5 and IKZF mutations (30). Nevertheless, our results were consistent, to a certain extent, with previous reports from China. For instance, Zheng et al. reported that FLT3 and TP53 mutations are common in Chinese ALL patients (31), and Zhang et al. reported high rates of FLT3 mutations in B-ALL (32). In contrast, a lower rate of FLT3 mutations (4–5%) has been reported in international studies (33, 34). In our study, all TP53 mutant chromosomes had a normal karyotype, although previous studies have shown that TP53 mutations usually occurred in hypodiploidy. All these differences may be related to differences in ethnicities as well as the number of cases.]. NOTCH1mutations are typically observed in T-ALL. However, studies have shown that NOTCH signaling receptors are also present on the surface of B cells (35), and NOTCH signaling has been implicated in B-ALL resistance (36).In our study, NOTCH1mutations were detected in 9.8% of B-ALL cases, and all 4 NOTCH1+ B-ALL cases were in the high-risk group, suggesting that NOTCH1 mutations may be associated with drug resistance and a poor prognosis in B-ALL.
The 5 genes with the highest mutation frequencies were analyzed for their correlation with prednisone sensitivity. TTN mutations as well as NOTCH1 mutations were found to show a high frequency in both B-ALL and T-ALL. Both these mutations were associated with resistance in the prednisone test. To our knowledge, the association between TTN mutation and glucocorticoid resistance has not been reported previously. While some studies have explored the relationship between Notch1 signaling and glucocorticoid resistance in T-ALL (37–41). However, these studies were largely performed in cell lines or animal models. Further, in studies examining clinical samples, the sample size was very limited. Therefore, our NOTCH1 mutation data are notable because our study is the first to report results from a large number of clinical samples. CREBBP and NT5C1 mutations, which have previously been linked to glucocorticoid resistance, were not detected in our study. TP53, MUC16, and FLT3 mutations showed no correlation with prednisone resistance, although statistical analysis revealed that TTN and MUC16 mutations were associated with a lower age among pediatric ALL patients.
The TTN gene (Titin, myonectin) is located on chromosome 2 at 2q31.2 and encodes a striated muscle protein. TTN mutations are associated with neuromuscular disease, cardiomyopathy, and the development of solid tumors (42). In terms of drug resistance, TTN mutations are associated with insulin tolerance in metabolic disease (43, 44) and the degree of anthracycline-induced myocardial damage (45). Lips et al. found that TTN is associated with chemotherapeutic drug tolerance in breast cancer (46, 47). Further, Jia et al. found TTN mutations in various solid tumors, such as breast cancer, lung cancer, and cervical cancer, and showed that these mutations were correlated with treatment response and prognosis (48). However, reports of TTN mutations in hematological tumors are currently rare. Skoczen et al. reported a TTN mutation in 1 patient with ALL and Netherton syndrome (49). Hence, although TTN mutations may be associated with glucocorticoid resistance in ALL, further data and research are required to validate this finding. The frequency of MUC16 mutations was also higher in the prednisone resistance group, although the difference was not statistically significant. The MUC16 (Mucin16) gene encodes a protein belonging to the mucin family, which is thought to be involved in barrier formation, protecting epithelial cells from pathogens (50). The product of this gene has been used as a marker for different cancers, and its expression is associated with a poor prognosis (51, 52). However, this mutation has not been reported in ALL. Additionally, our data analysis also revealed that NOTCH1 was associated with glucocorticoid resistance, consistent with previous reports of the link between mutations in this gene and a poor prognosis (53, 54).
Conclusions
The clinical features and gene mutation profiles of 71 pediatric ALL patients were obtained and investigated in the present study. We analyzed the differences in clinical features between pediatric ALL cases from our center and those examined in previous studies and described the common gene mutations as well as signaling pathways involved in pediatric ALL. Of note, our study is the first to report the association between TTN and NOTCH1 mutations and a low age of onset and glucocorticoid resistance in ALL. These findings could guide strategies for overcoming drug resistance and aid in the development of drug targets. However, owing to the small sample size of our study, more basic research and clinical studies examining a larger number of cases are required to validate our findings.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found here: NCBI Genebank, Accession ID PRJNA818462.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee Board of the Guangdong Provincial People's Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
JZ designed the study, carried out the statistical analysis and provided the data and pictures, drafted the initial manuscript, and approved the final manuscript as submitted. LZ and YW provided the chromosomal, FISH and RT-PCR information and approved the final manuscript as submitted. JP, BF, XL, and QY collected the clinical information for all the patients, and approved the final manuscript as submitted. All authors read and approved the final manuscript.
Funding
This work was supported by funding from the Guangdong Basic and Applied Basic Research Foundation (Grant Number 2018A030313524).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Special thank to all authors for their hard work.
Abbreviations
ALL, Acute lymphoblastic leukemia; OS, Overall survival; CNS, Central nervous system; FISH, Fluorescence in situ hybridization; WBC, White blood cell; LDH, Lactate dehydrogenase; WES, Whole exome sequencing; MICM, Morphology, immunology, cytology and molecular biology; TKI, Tyrosine kinase inhibitor; MRD, Minimal residual disease; Ph+, Philadelphia chromosome-positive.
Footnotes
References
1. PDQ Pediatric Treatment Editorial Board. Childhood Acute Lymphoblastic Leukemia Treatment (PDQ®): Health Professional Version; 2021. In: PDQ Cancer Information Summaries. Bethesda (MD): National Cancer Institute (US) (2002).
2. Inaba H, Mullighan CG. Pediatric acute lymphoblastic leukemia. Haematologica. (2020) 105:2524–39. doi: 10.3324/haematol.2020.247031
3. Bhatla T, Jones CL, Meyer JA, Vitanza NA, Raetz EA, Carroll WL. The biology of relapsed acute lymphoblastic leukemia: opportunities for therapeutic interventions. J Pediatr Hematol Oncol. (2014) 36:413–8. doi: 10.1097/MPH.0000000000000179
4. Scrideli CA, de Paula Queiróz R, Bernardes JE, Defavery R, Valera ET, Tone LG. Use of simplified strategies to evaluate early treatment response in childhood acute lymphoblastic leukemia. Leuk Res. (2006) 30:1049–52. doi: 10.1016/j.leukres.2005.11.021
5. Stanulla M, Cavé H, Moorman AV. IKZF1 deletions in pediatric acute lymphoblastic leukemia: still a poor prognostic marker? Blood. (2020) 135:252–60. doi: 10.1182/blood.2019000813
6. Xu N, Li YL Li X, Zhou X, Cao R, Li H, et al. Correlation between deletion of the CDKN2 gene and tyrosine kinase inhibitor resistance in adult Philadelphia chromosome-positive acute lymphoblastic leukemia. J Hematol Oncol. (2016) 9:40. doi: 10.1186/s13045-016-0270-5
7. Nicolaides NC, Charmandari E. Glucocorticoid Resistance. Exp Suppl. (2019) 111: 85–102. doi: 10.1007/978-3-030-25905-1_6
8. Hunger SP, Mullighan CG. Acute Lymphoblastic Leukemia in Children. N Engl J Med. (2015) 373:1541–52. doi: 10.1056/NEJMra1400972
9. Friedmann AM, Weinstein HJ. The role of prognostic features in the treatment of childhood acute lymphoblastic leukemia. Oncologist. (2000) 5:321–8. doi: 10.1634/theoncologist.5-4-321
10. Pui CH. Precision medicine in acute lymphoblastic leukemia. Front Med. (2020) 14:689–700. doi: 10.1007/s11684-020-0759-8
11. Cui L, Zhang RD, Gao C, Li WJ, Zhao XX, Zheng HY Li ZG, et al. [Evaluation of early response to treatment and its prognostic value in childhood acute lymphoblastic leukemia]. Zhongguo Shi Yan Xue Ye Xue Za Zhi. (2014) 22:298–303. doi: 10.7534/j.issn.1009-2137.2014.02.007
12. Coustan-Smith E, Sancho J, Hancock ML, Boyett JM, Behm FG, Raimondi SC, et al. Clinical importance of minimal residual disease in childhood acute lymphoblastic leukemia. Blood. (2000) 96:2691–6. doi: 10.1182/blood.V96.8.2691
13. Olivas-Aguirre M, Torres-López L, Pottosin I, Dobrovinskaya O. Overcoming glucocorticoid resistance in acute lymphoblastic leukemia: repurposed drugs can improve the protocol. Front Oncol. (2021) 11:617937. doi: 10.3389/fonc.2021.617937
14. Schmidt S, Rainer J, Ploner C, Presul E, Riml S, Kofler R. Glucocorticoid-induced apoptosis and glucocorticoid resistance: molecular mechanisms and clinical relevance. Cell Death Differ. (2004) 11:S45–55. doi: 10.1038/sj.cdd.4401456
15. Liu H, Li Z, Qiu F, Li C, Lin X, He Y, et al. Association Between NR3C1 Mutations and Glucocorticoid Resistance in Children With Acute Lymphoblastic Leukemia. Front Pharmacol. (2021) 12:634956. doi: 10.3389/fphar.2021.634956
16. Casale F, Addeo R, D'Angelo V, Indolfi P, Poggi V, Morgera C, et al. Determination of the in vivo effects of prednisone on Bcl-2 family protein expression in childhood acute lymphoblastic leukemia. Int J Oncol. (2003) 22:123–8. doi: 10.3892/ijo.22.1.123
17. Delgado-Martin C, Meyer LK, Huang BJ, Shimano KA, Zinter MS, Nguyen JV, et al. JAK/STAT pathway inhibition overcomes IL7-induced glucocorticoid resistance in a subset of human T-cell acute lymphoblastic leukemias. Leukemia. (2017) 31:2568–76. doi: 10.1038/leu.2017.136
18. De Smedt R, Morscio J, Goossens S, Van Vlierberghe P. Targeting steroid resistance in T-cell acute lymphoblastic leukemia. Blood Rev. (2019) 38:100591. doi: 10.1016/j.blre.2019.100591
19. Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet. (2013) 381:1943–55. doi: 10.1016/S0140-6736(12)62187-4
20. Zhou Y, You MJ, Young KH, Lin P, Lu G, Medeiros LJ, et al. Advances in the molecular pathobiology of B-lymphoblastic leukemia. Hum Pathol. (2012) 43:1347–62. doi: 10.1016/j.humpath.2012.02.004
21. Ferrando AA, Look AT. Gene expression profiling in T-cell acute lymphoblastic leukemia. Semin Hematol. (2003) 40:274–80. doi: 10.1016/S0037-1963(03)00195-1
22. Lee DS, Kim YR, Cho HK, Lee CK, Lee JH, Cho HI. The presence of TEL/AML1 rearrangement and cryptic deletion of the TEL gene in adult acute lymphoblastic leukemia (ALL). Cancer Genet Cytogenet. (2005) 162:176–8. doi: 10.1016/j.cancergencyto.2005.02.020
23. Aydin C, Cetin Z, Manguoglu AE, Tayfun F, Clark OA, et al. Evaluation of ETV6/RUNX1 fusion and additional abnormalities involving ETV6 and/or RUNX1 genes using fish technique in patients with childhood acute lymphoblastic leukemia. Indian J Hematol Blood Transfus. (2016) 32:154–61. doi: 10.1007/s12288-015-0557-7
24. Zuna J, Zaliova M, Muzikova K, Meyer C, Lizcova L, Zemanova Z, et al. Acute leukemias with ETV6/ABL1 (TEL/ABL) fusion: poor prognosis and prenatal origin. Genes Chromosomes Cancer. (2010) 49:873–84. doi: 10.1002/gcc.20796
25. Zhou F, Wen Y, Jin R, Chen H. New attempts for central nervous infiltration of pediatric acute lymphoblastic leukemia. Cancer Metastasis Rev. (2019) 38:657–71. doi: 10.1007/s10555-019-09827-z
26. Jeha S, Pei D, Choi J, Cheng C, Sandlund JT, Coustan-Smith E, et al. Improved CNS Control of Childhood Acute Lymphoblastic Leukemia Without Cranial Irradiation: St Jude Total Therapy Study 16. J Clin Oncol. (2019) 37:3377–91. doi: 10.1200/JCO.19.01692
27. Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. (2017) 49:1211–8. doi: 10.1038/ng.3909
28. Shen Z, Chu XL, Wang RX Li JL, Liu MY, Xie YY, et al. The clinical and molecular characteristics of FLT3 mutations in chinese de novo adolescent and adult acute lymphoblastic leukemia patients. Clin Lymphoma Myeloma). (2020) 20:e259–69. doi: 10.1016/j.clml.2019.09.602
29. Karabacak BH, Erbey F, Bayram I, Yilmaz S, Acipayam C, Kilinç Y, Tanyeli A. Fms-like tyrosine kinase 3 mutations in childhood acute leukemias and their association with prognosis. Asian Pac J Cancer. (2010) 11:923–7.
30. Mullighan CG. Genomic profiling of B-progenitor acute lymphoblastic leukemia. Best Pract Res Clin Haematol. (2011) 24:489–503. doi: 10.1016/j.beha.2011.09.004
31. Zheng RY, Wang SJ, Wang C, Li T, Liao LX, Li ML, et al. [Gene Mutation in Acute Lymphoblastic Leukemia by DNA Sequencing]. J Exp Hematol. (2020) 28:1791–5.
32. Zhang H, Wang H, Qian X, Gao S, Xia J, Liu J, et al. Genetic mutational analysis of pediatric acute lymphoblastic leukemia from a single center in China using exon sequencing. BMC Cancer. (2020) 20:211. doi: 10.1186/s12885-020-6709-7
33. Alkhayat N, Elborai Y, Al Sharif O, Al Shahrani M, Alsuhaibani O, Awad M, et al. Cytogenetic profile and FLT3 gene mutations of childhood acute lymphoblastic leukemia. Clin Med Insights Oncol. (2017) 11:1179554917721710. doi: 10.1177/1179554917721710
34. Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. (2007) 446:758–64. doi: 10.1038/nature05690
35. Nwabo Kamdje AH, Krampera M. Notch signaling in acute lymphoblastic leukemia: any role for stromal microenvironment? Blood. (2011) 118:6506–14. doi: 10.1182/blood-2011-08-376061
36. Takam Kamga P, Dal Collo G, Midolo M, Adamo A, Delfino P, Mercuri A, et al. Inhibition of notch signaling enhances chemosensitivity in B-cell precursor acute lymphoblastic leukemia. Cancer Res. (2019) 79:639–49. doi: 10.1158/0008-5472.CAN-18-1617
37. Franciosa G, Smits JGA, Minuzzo S, Martinez-Val A, Indraccolo S, Olsen JV. Proteomics of resistance to Notch1 inhibition in acute lymphoblastic leukemia reveals targetable kinase signatures. Nat Commun. (2021) 12:2507. doi: 10.1038/s41467-021-22787-9
38. Real PJ, Tosello V, Palomero T, Castillo M, Hernando E, de Stanchina E, et al. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med. (2009) 15:50–8. doi: 10.1038/nm.1900
39. Real PJ, Ferrando AA. NOTCH inhibition and glucocorticoid therapy in T-cell acute lymphoblastic leukemia. Leukemia. (2009) 23:1374–7. doi: 10.1038/leu.2009.75
40. Zhu YM, Zhao WL, Fu JF, Shi JY, Pan Q, Hu J, et al. NOTCH1 mutations in T-cell acute lymphoblastic leukemia: prognostic significance and implication in multifactorial leukemogenesis. Clin Cancer Res. (2006) 12:3043–9. doi: 10.1158/1078-0432.CCR-05-2832
41. uurbier L, Homminga I, Calvert V, te Winkel ML, Buijs-Gladdines JG, Kooi C, et al. NOTCH1 and/or FBXW7 mutations predict for initial good prednisone response but not for improved outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on DCOG or COALL protocols. Leukemia. (2010) 24:2014–22. doi: 10.1038/leu.2010.204
42. Kellermayer D, Smith JE 3rd, Granzier H. Titin mutations and muscle disease. Pflugers. (2019) 471:673–82. doi: 10.1007/s00424-019-02272-5
43. Li S, Liang M, Gao D, Su Q, Laher I. Changes in titin and collagen modulate effects of aerobic and resistance exercise on diabetic cardiac function. J Cardiovasc Transl Res. (2019) 12:404–14. doi: 10.1007/s12265-019-09875-4
44. Durak A, Bitirim CV, Turan B. Titin and CK2α are New Intracellular Targets in Acute Insulin Application-Associated Benefits on Electrophysiological Parameters of Left Ventricular Cardiomyocytes from Insulin-Resistant Metabolic Syndrome. Cardiovasc Drugs. (2020) 34:487–501. doi: 10.1007/s10557-020-06974-2
45. Sawyer DB, Peng X, Chen B, Pentassuglia L, Lim CC. Mechanisms of anthracycline cardiac injury: can we identify strategies for cardioprotection? Prog Cardiovasc Dis. (2010) 53:105–13. doi: 10.1016/j.pcad.2010.06.007
46. Lips EH, Michaut M, Hoogstraat M, Mulder L, Besselink NJ, Koudijs MJ, et al. Next generation sequencing of triple negative breast cancer to find predictors for chemotherapy response. Breast Cancer Res. (2015) 17:134. doi: 10.1186/s13058-015-0642-8
47. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. (2013) 499:214–8. doi: 10.1038/nature12213
48. Jia Q, Wang J, He N, He J, Zhu B. Titin mutation associated with responsiveness to checkpoint blockades in solid tumors. JCI Insight. (2019) 4:e127901. doi: 10.1172/jci.insight.127901
49. Skoczen S, Stepien K, Mlynarski W, Centkowski P, Kwiecinska K, Korostynski M, et al. Genetic signature of acute lymphoblastic leukemia and Netherton syndrome co-incidence-first report in the literature. Front Oncol. (2020) 9:1477. doi: 10.3389/fonc.2019.01477
50. Ma J, Rubin BK, Voynow JA. Mucins, Mucus, and Goblet Cells. Chest. (2018) 154:169–76. doi: 10.1016/j.chest.2017.11.008
51. Felder M, Kapur A, Gonzalez-Bosquet J, Horibata S, Heintz J, Albrecht R, et al. MUC16 (CA125): tumor biomarker to cancer therapy, a work in progress. Mol Cancer. (2014) 13:129. doi: 10.1186/1476-4598-13-129
52. Aithal A, Rauth S, Kshirsagar P, Shah A, Lakshmanan I, Junker WM, et al. MUC16 as a novel target for cancer therapy. Expert Opin Ther Targets. (2018) 22:675–86. doi: 10.1080/14728222.2018.1498845
53. Follini E, Marchesini M, Roti G. Strategies to overcome resistance mechanisms in T-cell acute lymphoblastic leukemia. Int J Mol Sci. (2019) 20:3021. doi: 10.3390/ijms20123021
Keywords: TTN, NOTCH1, gene mutation, acute lymphoblastic leukemia, drug resistance
Citation: Zhang J, Zeng L, Wang Y, Pan J, Li X, Feng B and Yang Q (2022) Gene Mutations Related to Glucocorticoid Resistance in Pediatric Acute Lymphoblastic Leukemia. Front. Pediatr. 10:831229. doi: 10.3389/fped.2022.831229
Received: 08 December 2021; Accepted: 13 May 2022;
Published: 06 June 2022.
Edited by:
Jutte Van Der Werff Ten Bosch, University Hospital Brussels, BelgiumReviewed by:
Yongsheng Ruan, Southern Medical University, ChinaJennifer McNeer, The University of Chicago, United States
Copyright © 2022 Zhang, Zeng, Wang, Pan, Li, Feng and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: JinFang Zhang, amluZmFuZ3poYW5nMTIxNkAxMjYuY29t