 Jaewon Kim
Jaewon Kim Jaewoong Lee
Jaewoong Lee Dae-Hyun Jang
Dae-Hyun Jang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 18 May 2022
Sec. Genetics of Common and Rare Diseases
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.812408
This article is part of the Research Topic Inherited Metabolic Diseases in Pediatrics: Clinical and Molecular Features View all 25 articles
Leigh syndrome is a neurodegenerative disorder that presents with fluctuation and stepwise deterioration, such as neurodevelopmental delay and regression, dysarthria, dysphagia, hypotonia, dystonia, tremor, spasticity, epilepsy, and respiratory problems. The syndrome characteristically presents symmetric necrotizing lesions in the basal ganglia, brainstem, cerebellum, thalamus, and spinal cord on cranial magnetic resonance imaging. To date, more than 85 genes are known to be associated with Leigh syndrome. Here, we present a rare case of a child who developed Leigh syndrome due to pathogenic variants of NDUFAF6, which encodes an assembly factor of complex I, a respiratory chain subunit. A targeted next-generation sequencing analysis related to mitochondrial disease revealed a missense variant (NM_152416.4:c.371T > C; p.Ile124Thr) and a frameshift variant (NM_152416.4:c.233_242del; p.Leu78GInfs*10) inherited biparentally. The proband underwent physical therapy and nutrient cocktail therapy, but his physical impairment gradually worsened.
Mitochondrial disease is a common inherited metabolic disorder that is clinically and genetically heterogeneous and occurs in approximately 10–25 cases per 100,000 individuals, many of which are childhood-onset usually by the age of 3 years (1). Mitochondria are present in all cells except erythrocytes and, given that mitochondria are responsible for oxidative phosphorylation and ATP synthesis, mitochondrial disease exhibits diverse phenotypes depending on the affected organs. Mitochondrial dysfunction leads to multiple system disorder, manifesting a progressive course with high morbidity and mortality (2). Mitochondrial disease is a genetic disorder resulting from pathogenic variants of mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) associated with mitochondrial function. The mtDNA consists of 37 genes and encodes 13 proteins, and over 1,000 nuclear genes encode proteins related to mitochondrial function (3).
The most common mitochondrial disease that occurs during childhood is Leigh syndrome, with a prevalence of approximately 2.5 cases per 100,000 individuals (4). Most cases of Leigh syndrome occur in childhood and rarely in adolescence or early adulthood. Leigh syndrome is a neurodegenerative disorder, with characteristic symmetric, necrotizing lesions in the basal ganglia, brainstem, cerebellum, thalamus, and spinal cord on cranial magnetic resonance imaging (MRI). The phenotypes of Leigh syndrome include neurological symptoms showing fluctuation and stepwise deterioration, such as neurodevelopmental delay and regression, dysarthria, dysphagia, hypotonia, dystonia, tremor, spasticity, epilepsy, and respiratory problems. Other non-neurologic symptoms include cardiac abnormalities, hepatorenal anomalies, anemia, and gastrointestinal symptoms such as constipation and vomiting (5). More than 85 genes are related to Leigh syndrome including mtDNA and nDNA, and genes related to complex I (NADH: ubiquinone oxidoreductase) deficiency are the most common causes. Approximately 75–80% of cases of Leigh syndrome are caused by pathogenic nDNA variants and more than half of the nDNA genes are related to complex I. NDUFAF6 (NADH: ubiquinone oxidoreductase complex assembly factor 6) is one of them, and NDUFAF6 is one of the major genes that cause reduced complex I activity by mitochondrial respiratory chain complex defect. Biallelic missense variants of NDUFAF6 are known to cause Leigh syndrome.
Here, we present a rare case of a child who developed Leigh syndrome due to pathogenic variants of NDUFAF6 on chromosome 8, which encodes an assembly factor of complex I, a respiratory chain subunit. A targeted next-generation sequencing study (NGS) enabled rapid genetic confirmation and avoided invasive diagnostic procedures such as skeletal muscle biopsy and skin fibroblast cell culture.
A 56-month-old boy was referred to a rehabilitation clinic due to left hand clumsiness and frequent falling. The symptoms gradually began around the age of 3 years and slowly progressed. The patient had been born after an uncomplicated 40-week gestation with a birth weight of 3.07 kg. There were no perinatal problems and no specific family history of interest. There was no psychomotor developmental abnormalities prior to the onset of symptoms. The patient was brought to the primary clinic at 55 months due to language developmental delay, with simplified sentences and inaccurate pronunciation. In the evaluation, the patient’s overall comprehension and language expression level was measured around 56–60 months, and his consonant accuracy was evaluated at approximately 87% in the pronunciation intelligibility assessment. The patient also presented with weakened tongue movement, simplification of middle consonants, and simplification of liquid consonants. In the psychoeducational profile–revised (PEP-R) evaluation, the overall developmental level was 64–65 months, and the other domains were above the normal ranges, except for fine motor function, which was below the average of 52–54 months. During the examination, the patient presented poor bimanual manipulation, with left hand clumsiness and slight weakness of the right ankle dorsiflexion; the patient was referred to a tertiary medical center.
During the physical examination, the patient presented with dystonic and ataxic movements on the left side and clumsy movements of both hands. Weakness in ankle dorsiflexion involving both sides and spastic hypertonia in the left upper and lower extremities were observed. The strength of both ankle dorsiflexors was 3/5, and the strength of the other muscles was 5/5. The patient had difficulty jumping on both feet and stair-climbing and descending. Poor balance with one-leg standing with eyes open was also observed. The patient could independently walk but had tip-toeing and limping gait patterns. In the functional assessment, the balance evaluation using the Berg balance scale (BBS) revealed a poor standing balance of 36/56. The total score on the gross motor function measure-88 (GMFM-88) was 81.82%. The Beery–Buktenica developmental test of visual motor integration (6th edition) revealed a normal range of visual motor function of 99 (47%) with impaired motor coordination ability of 73 (4%).
Due to a suspected brain pathology, an MRI of the head was performed, revealing abnormal signals in the bilateral putamen, with slight volume reduction and partial necrosis. The MRI revealed high signal intensity on T2- and diffusion-weighted images, an apparent diffusion coefficient (ADC), and fluid-attenuated inversion recovery (FLAIR) images with low signal intensity on T1-weighted images in the bilateral putamen (Figure 1). MR angiography showed normal cerebral vessels. To differentiate diseases that could show lesions of symmetric basal ganglia, we also performed MR spectroscopy, detecting normal N-acetylaspartate, choline, and creatine levels, and increased lipid and lactate levels (Figure 2). The high lactate peak would indicate acute mitochondrial dysfunction and with clinical features, we determined that there was a possibility of mitochondrial encephalopathy, especially Leigh syndrome. The laboratory test revealed a lactic acid level of 2.45 mmol/L (range, 0.50–2.20 mmol/L) and a creatine phosphokinase level of 287 U/L (range, 0–250 U/L). In the urine organic acid test, a slight increase was observed in tiglylglycine levels to 0.4 mmol/mol creatine (range, 0–0.2 mmol/mol creatine). The other laboratory test results, including serum aspartate aminotransferase, alanine aminotransferase, amino acid analysis, and acylcarnitine were all within normal ranges.
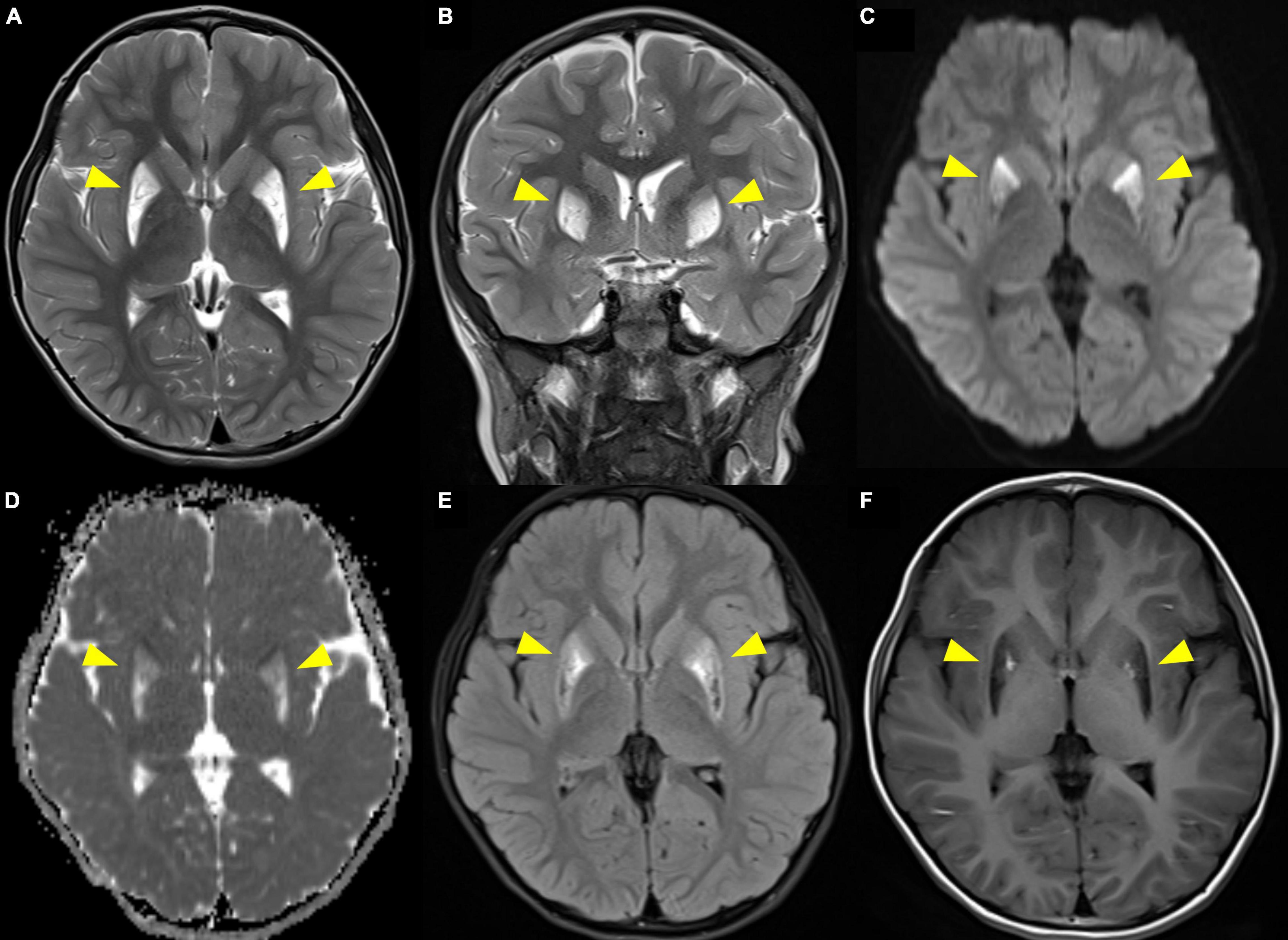
Figure 1. Brain MRI showing high signal intensity on T2-weighted imaging (A. axial, B. coronal) and diffusion-weighted imaging (C), apparent diffusion coefficient (D), and fluid-attenuated inversion recovery (E) images with low signal intensity on T1-weighted imaging (F) in the bilateral putamen. Arrowheads indicate the lesions.
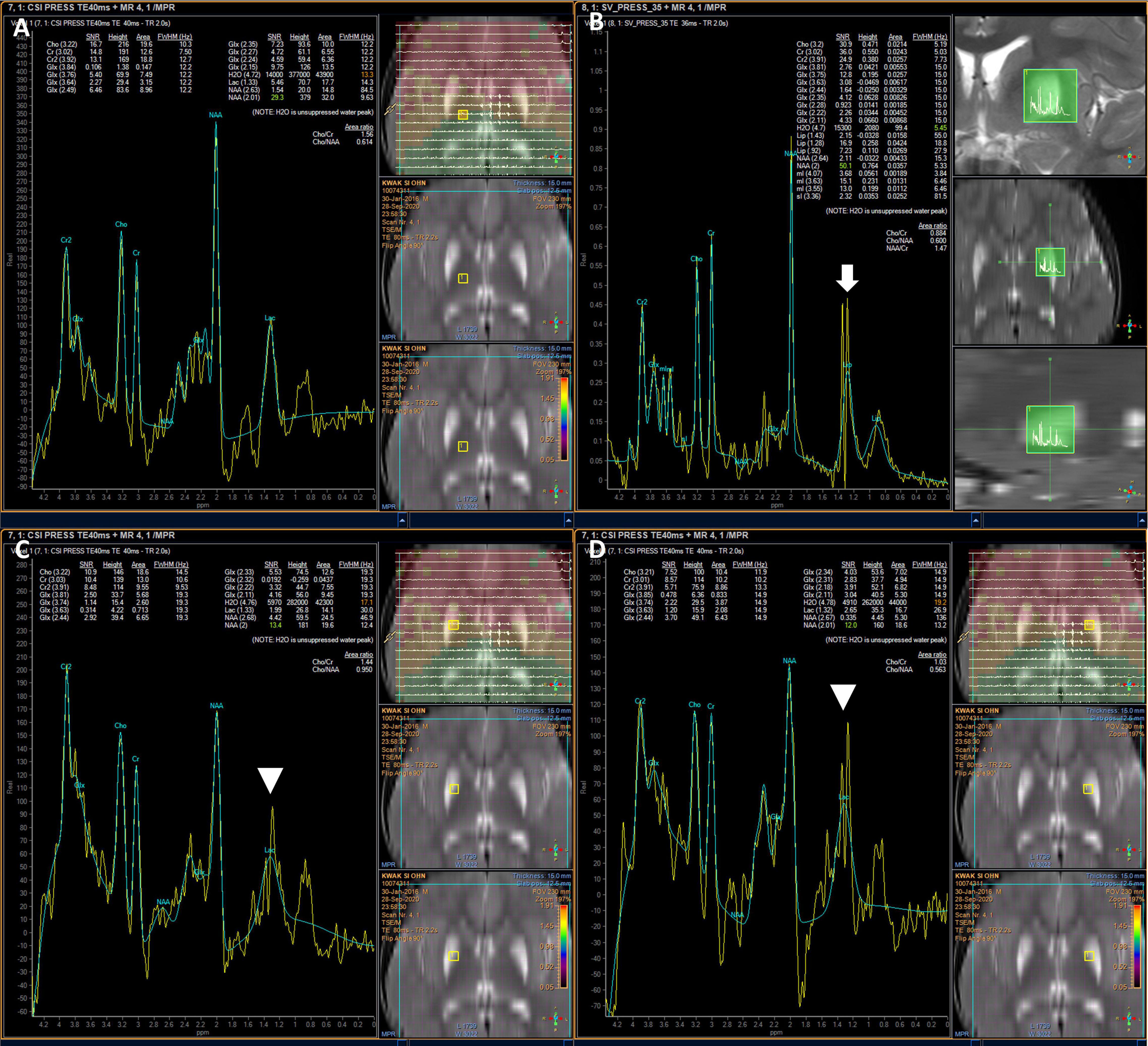
Figure 2. Brain MR spectroscopy showing increased lipid and lactate in the lesions. Normal patterns outside lesions (A), elevated lipids (white arrow) (B), and elevated lactate in both putamen (arrowhead) (C,D).
We performed a mitochondrial DNA sequencing test and targeted NGS analysis of a multi-gene panel related to the mitochondrial disease. Informed consent from the patient’s guardian was collected. Libraries were prepared with the Illumina™ Truseq DNA kit, and sequencing was performed using the Illumina™ Nextseq platform. The whole mtDNA test revealed a normal mtDNA sequence. The NGS test revealed two heterozygous variants of NDUFAF6 in the different exons, identifying a missense variant (NM_152416.4:c.371T > C; p.Ile124Thr) and a frameshift variant (NM_152416.4:c.233_242del; p.Leu78GInfs*10), which were validated by Sanger sequencing. No other significant variants were found. The missense variant c.371T > C has been previously reported to cause Leigh syndrome, while the c.233_242del variant has not been reported (6–8). The missense variant c.371T > C was classified as a likely pathogenic variant and the frameshift variant (c.233_242del) was classified as a pathogenic variant by American College of Medical Genetics and Genomics (ACMG) Classification Standards and Guidelines for Genetic Variations. In silico prediction for the missense variant, SIFT predicted as deleterious (score: 0.05), PolyPhen-2 as benign (score 0.255), and MutationTaster as disease causing (probability: 0.99). For the frameshift variant, MutationTaster predicted as disease causing (probability: 1). To identify these compound heterozygous variants, we performed targeted gene sequencing of the parents, which revealed that the c.371T > C variant was inherited from the mother while the c.233_242del variant was inherited from the father (Figure 3). Finally, the proband was diagnosed with Leigh syndrome caused by biallelic pathogenic variants of NDUFAF6, inherited biparentally.
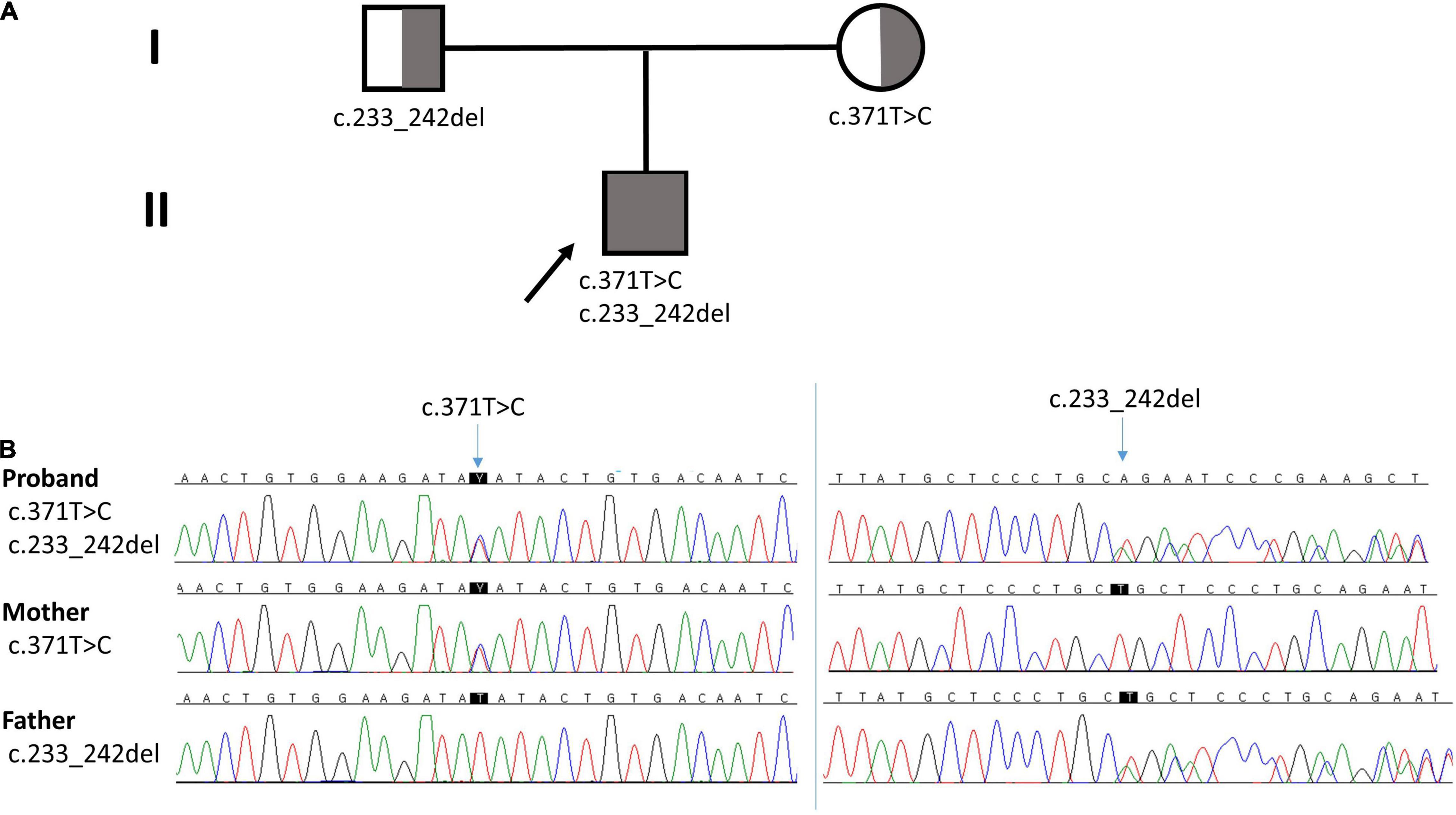
Figure 3. (A) Pedigree of the family. Arrow indicates the proband. (B) Chromatographs of NDUFAF6. The proband had a missense variant (c.371T > C) from the mother and a frameshift variant (c.233_242del) from the father.
After the diagnosis, the patient continued physiotherapy and occupational therapy while undergoing nutrient cocktail therapy. He was prescribed 10 mg of biotin, 500 mg of thiamine, 100 mg of Coenzyme Q10, and 100 mg of L-carnitine per day. However, worsening and improvement of spastic dystonia and balance impairment were repeated and his gait instability gradually worsened.
Five months after the first visit and at the age of 61 months, the patient’s BBS dropped from 36/56 to 11/56, and his GMFM-88 score worsened from 81.82 to 58.33%. He required support from two hands for walking and for transitioning from sitting to standing. He was unable to maintain an upright sitting or standing position and showed poor sitting balance. A symmetric tonic neck reflex appeared, and a hand-foot crawling pattern was observed.
We have reported a proband with biallelic pathogenic variants, c.371T > C and c.233_242del in NDUFAF6, who showed the typical phenotype of Leigh syndrome. The proband presented Leigh syndrome-appropriate clinical features, brain MRI and MR spectroscopy findings, and genetic confirmation. However, the serum biomarkers showing oxidative phosphorylation impairment such as lactate, pyruvate, creatine kinase, amino acid, acylcarnitine, and organic acid revealed no disease-specific findings. The patient underwent rehabilitation therapy and nutrient cocktail therapy, but his physical impairment rapidly worsened.
NDUFAF6 is an assembly factor of NADH-ubiquinone oxidoreductase (complex 1) and plays an important role in complex 1 maturation and activity. NDUFAF6 is a 38 kDa protein, consists of 333 amino acids, and is involved in the assembly of complex I in early stages, playing a role in regulating and stabilizing the complex I subunit MT-ND1. Defects in NDUFAF6 isoform 1 can disrupt complex I assembly by causing rapid proteolysis of the newly formed ND1 subunit, adversely affecting mitochondrial function (9, 10). Biallelic pathogenic variants cause Leigh syndrome, and 16 variants have been reported in 20 patients to date (Table 1 and Figure 4). Among them, the most variants have been reported in exon 2, and 2 variants have also been reported in exons 3 and 5. All children with NDUFAF6-related Leigh syndrome show necrotic lesions in the symmetric basal ganglia, dystonia, and neurological regression. Characteristically, NDUFAF6-related Leigh syndrome tends to show normal or slight higher serum lactate, and our proband also showed lactate close to the normal range, revealing little diagnostic value for Leigh syndrome.
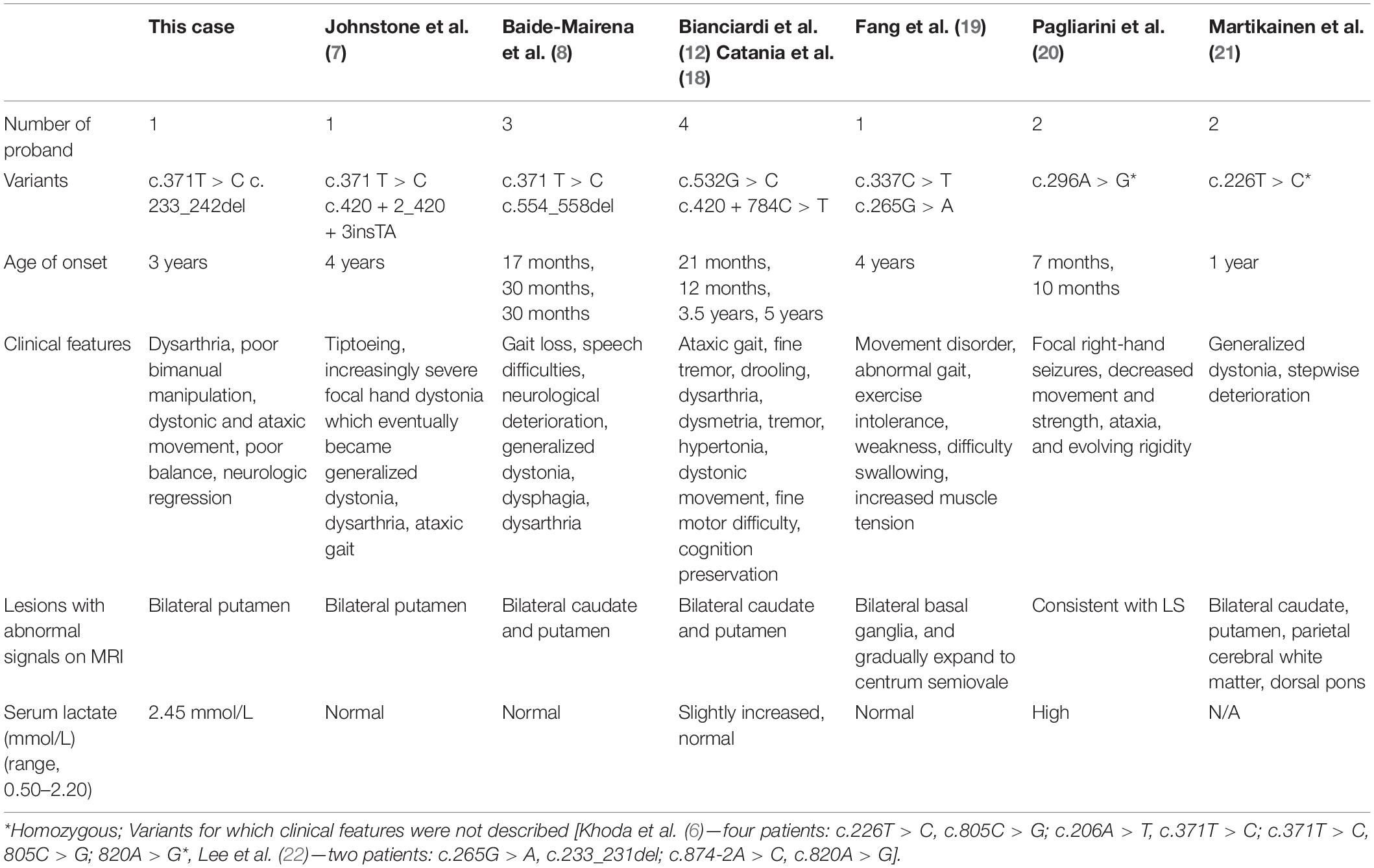
Table 1. Variants and clinical features of patients with NDUFAF6-related Leigh syndrome.
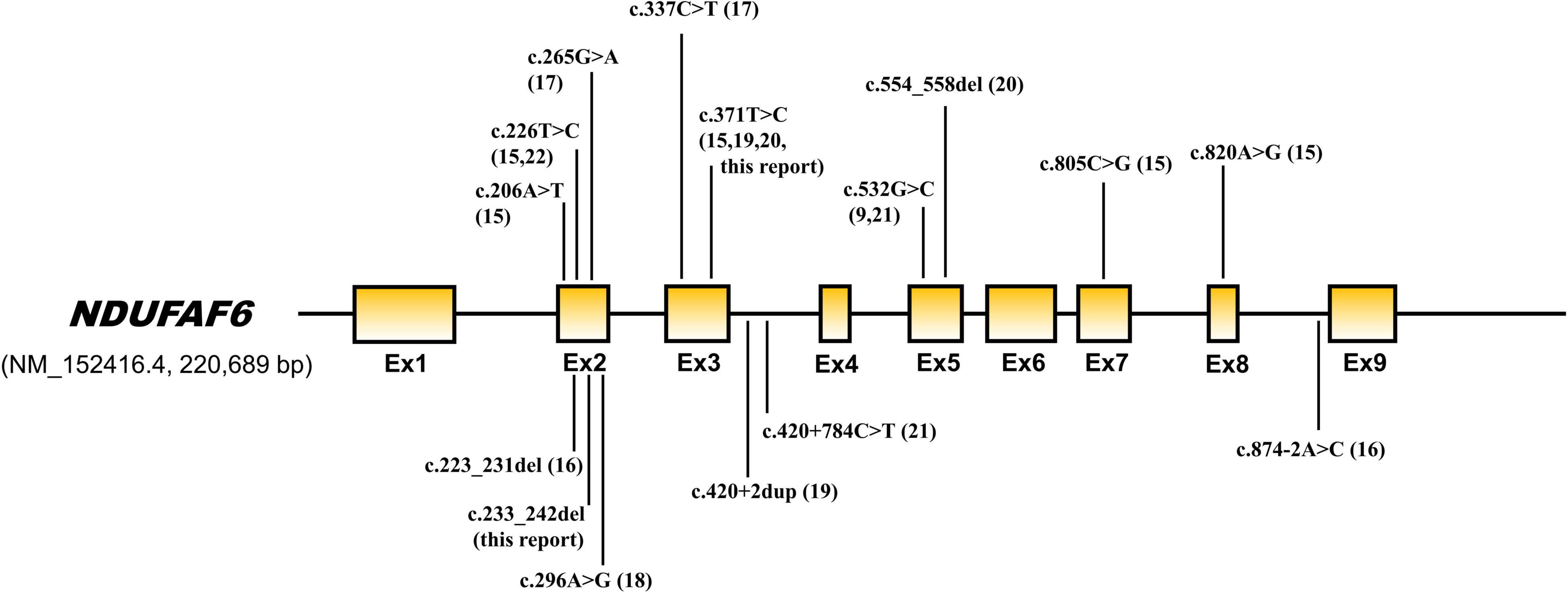
Figure 4. Genetic structure of NDUFAF6. Pathogenic variants previously reported are indicated by arrows.
In a cohort study of 166 patients with Leigh syndrome, 8 had pathogenic variants of NDUFAF6. As a result of the analysis, mtDNA variants were associated with high mortality, and patients with NDUFAF6 variants had better outcomes than those with other nDNA gene variants showing lower mortality and longer motor function preservation (11). However, not all patients with NDUFAF6-related Leigh syndrome show mild phenotypes, and the genotype does not appear to predict the prognosis sufficiently, given that there are patients with early disease onset with severe symptoms who have the same variants (12). Our proband showed normal psychomotor development with uneventful medical history until the age of 3, and symptoms developed gradually. At 55 months, he showed mild motor impairment, but from then on, he showed rapid deterioration. Since there is no report on long term functional prognosis in the NDUFAF6-related Leigh syndrome, additional reports will be helpful for prognosis prediction.
Mitochondrial disease is difficult to diagnose due to the diverse clinical features. Previously, tissue biopsy was often performed for diagnosis by histochemical and biochemical analysis. With the recent development of NGS technology, there have been numerous cases that relied on genetic tests. In addition, cases previously diagnosed with probable or uncertain mitochondrial disease have more frequently been genetically confirmed. As a result, overall diagnostic success has risen from less than 20% to more than 60% (1). In particular, given that 75–80% of cases of childhood-onset mitochondrial disease occur due to the pathogenic variant of nDNA, the NGS test increases the diagnosis success (13). In a study conducted in 2015, patients who were ultimately diagnosed with mitochondrial disease had met an average of 8.19 physicians before being confirmed, and 70% of the patients had undergone invasive muscle biopsy. Rapid diagnosis without invasive tests has been made possible with the advances in genetic diagnostic technology, including whole exome sequencing and whole genome sequencing (14).
Treatment for Leigh syndrome has no clear evidence, but cocktails of nutritional dietary supplements, such as antioxidants, vitamins, coenzyme Q10, and nitric acid precursors have been widely used (15). Given that Leigh syndrome is genetically heterogeneous, early intervention with tailored therapy according to the genotype rather than treatment according to the phenomenon will be an important treatment direction in the future. In terms of rehabilitation, exercise prescription including aerobic exercise training is often performed for patients with mitochondrial disease with gait disturbance, imbalance, frequent falling, fatigue, and exercise intolerance. In general, excessive physical activity is not recommended, and physiotherapy is contraindicated for patients with cardiac involvement. Physiotherapy is also not recommended for those with acute illness, severe fatigue, and a generally deconditioned status because of the expected excessive energy demand in addition to the body’s energy demands (16). However, there was a report that oxygen utilization, exercise tolerance, and quality of life were better for the group who continued exercise training than for the group who did not (17). Therefore, rehabilitation therapy should be determined individually. Our patient underwent rehabilitation therapy because his general condition was not poor and did not correspond to discontinuation; however, his physical function gradually deteriorated.
We report a rare genetic disorder: NDUFAF6-related Leigh syndrome resulting from biallelic pathogenic variants, c.371T > C and c.233_242del in NDUFAF6. The proband showed typical Leigh syndrome features, and his physical impairment gradually worsened despite several therapies. This case report is distinguished from those previously reported in that we describe in detail the clinical features, including the developmental evaluation and clinical progressions.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the Institutional Review Board for Clinical Research at Incheon St. Mary’s Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
JK: acquisition of data, analysis and interpretation of data, and writing. JL: analysis and interpretation of data. D-HJ: study concept and design, acquisition of data, analysis and interpretation of data, study supervision, and critical revision of manuscript for intellectual content. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, et al. Mitochondrial diseases. Nat Rev Dis Primers. (2016) 2:16080. doi: 10.1038/nrdp.2016.80
2. McFarland R, Taylor RW, Turnbull DM. A neurological perspective on mitochondrial disease. Lancet Neurol. (2010) 9:829–40.
3. Calvo SE, Mootha VK. The mitochondrial proteome and human disease. Annu Rev Genomics Hum Genet. (2010) 11:25–44.
4. Lake NJ, Compton AG, Rahman S, Thorburn DR. Leigh syndrome: one disorder, more than 75 monogenic causes. Ann Neurol. (2016) 79:190–203. doi: 10.1002/ana.24551
5. Gerards M, Sallevelt SC, Smeets HJ. Leigh syndrome: resolving the clinical and genetic heterogeneity paves the way for treatment options. Mol Genet Metab. (2016) 117:300–12. doi: 10.1016/j.ymgme.2015.12.004
6. Kohda M, Tokuzawa Y, Kishita Y, Nyuzuki H, Moriyama Y, Mizuno Y, et al. A comprehensive genomic analysis reveals the genetic landscape of mitochondrial respiratory chain complex deficiencies. PLoS Genet. (2016) 12:e1005679. doi: 10.1371/journal.pgen.1005679
7. Johnstone T, Wang J, Ross D, Balanda N, Huang Y, Godfrey R, et al. Biallelic variants in two complex I genes cause abnormal splicing defects in probands with mild leigh syndrome. Mol Genet Metab. (2020) 131:98–106. doi: 10.1016/j.ymgme.2020.09.008
8. Baide-Mairena H, Gaudó P, Marti-Sánchez L, Emperador S, Sánchez-Montanez A, Alonso-Luengo O, et al. Mutations in the mitochondrial complex I assembly factor NDUFAF6 cause isolated bilateral striatal necrosis and progressive dystonia in childhood. Mol Genet Metab. (2019) 126:250–8.
9. Andrews B, Carroll J, Ding S, Fearnley IM, Walker JE. Assembly factors for the membrane arm of human complex I. Proc Natl Acad Sci USA. (2013) 110:18934–9. doi: 10.1073/pnas.1319247110
10. Lemire BD. Evolution, structure and membrane association of NDUFAF6, an assembly factor for NADH: ubiquinone oxidoreductase (Complex I). Mitochondrion. (2017) 35:13–22. doi: 10.1016/j.mito.2017.04.005
11. Ogawa E, Fushimi T, Ogawa-Tominaga M, Shimura M, Tajika M, Ichimoto K, et al. Mortality of Japanese patients with Leigh syndrome: Effects of age at onset and genetic diagnosis. J Inherit Metab Dis. (2020) 43:819–26. doi: 10.1002/jimd.12218
12. Bianciardi L, Imperatore V, Fernandez-Vizarra E, Lopomo A, Falabella M, Furini S, et al. Exome sequencing coupled with mRNA analysis identifies NDUFAF6 as a leigh gene. Mol Genet Metab. (2016) 119:214–22. doi: 10.1016/j.ymgme.2016.09.001
13. Thorburn DR. Mitochondrial disorders: prevalence, myths and advances. J Inherit Metab Dis. (2004) 27:349–62.
14. Grier J, Hirano M, Karaa A, Shepard E, Thompson JLP. Diagnostic odyssey of patients with mitochondrial disease: results of a survey. Neurol Genet. (2018) 4:e230. doi: 10.1212/NXG.0000000000000230
15. Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF. Treatment for mitochondrial disorders. Cochrane Database Syst Rev. (2012) 2012:CD004426. doi: 10.1002/14651858.CD004426.pub3
16. Newcastle University. Physiotherapy Guidance for People with Mitochondrial Disease. (2021). Available online at: https://www.newcastle-mitochondria.com/clinical-professional-home-page/clinical-publications/clinical-guidelines/ (Accessed August 19, 2021)
17. Taivassalo T, Gardner JL, Taylor RW, Schaefer AM, Newman J, Barron MJ, et al. Endurance training and detraining in mitochondrial myopathies due to single large-scale mtDNA deletions. Brain. (2006) 129:3391–401. doi: 10.1093/brain/awl282
18. Catania A, Ardissone A, Verrigni D, Legati A, Reyes A, Lamantea E, et al. Compound heterozygous missense and deep intronic variants in NDUFAF6 unraveled by exome sequencing and mRNA analysis. J Hum Genet. (2018) 63:563–8. doi: 10.1038/s10038-018-0423-1
19. Fang F, Liu Z, Fang H, Wu J, Shen D, Sun S, et al. The clinical and genetic characteristics in children with mitochondrial disease in China. Sci China Life Sci. (2017) 60:746–57. doi: 10.1007/s11427-017-9080-y
20. Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. (2008) 134:112–23. doi: 10.1016/j.cell.2008.06.016
21. Martikainen MH, Ng YS, Gorman GS, Alston CL, Blakely EL, Schaefer AM, et al. Clinical, genetic, and radiological features of extrapyramidal movement disorders in mitochondrial disease. JAMA Neurol. (2016) 73:668–74. doi: 10.1001/jamaneurol.2016.0355
Keywords: Leigh syndrome, mitochondrial disease, NDUFAF6, complex I deficiency, neurodegenerative disorder
Citation: Kim J, Lee J and Jang D-H (2022) NDUFAF6-Related Leigh Syndrome Caused by Rare Pathogenic Variants: A Case Report and the Focused Review of Literature. Front. Pediatr. 10:812408. doi: 10.3389/fped.2022.812408
Received: 10 November 2021; Accepted: 31 March 2022;
Published: 18 May 2022.
Edited by:
Huiwen Zhang, Xinhua Hospital, ChinaReviewed by:
Juan Dario Ortigoza-Escobar, Hospital Sant Joan de Déu Barcelona, SpainCopyright © 2022 Kim, Lee and Jang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dae-Hyun Jang, ZGhqYW5nbWRAbmF2ZXIuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.