 Dhirendra K. Singh
Dhirendra K. Singh Claire M. Miller
Claire M. Miller Kelly A. Orgel1
Kelly A. Orgel1 Stephen Mackay
Stephen Mackay Misty Good
Misty Good
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr., 11 January 2023
Sec. Neonatology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.1107404
This article is part of the Research TopicRecent Advances in Our Understanding of NEC Pathogenesis, Diagnosis, and TreatmentView all 12 articles
Necrotizing enterocolitis (NEC) is a devastating, multifactorial disease mainly affecting the intestine of premature infants. Recent discoveries have significantly enhanced our understanding of risk factors, as well as, cellular and genetic mechanisms of this complex disease. Despite these advancements, no essential, single risk factor, nor the mechanism by which each risk factor affects NEC has been elucidated. Nonetheless, recent research indicates that maternal factors, antibiotic exposure, feeding, hypoxia, and altered gut microbiota pose a threat to the underdeveloped immunity of preterm infants. Here we review predisposing factors, status of unwarranted immune responses, and microbial pathogenesis in NEC based on currently available scientific evidence. We additionally discuss novel techniques and models used to study NEC and how this research translates from the bench to the bedside into potential treatment strategies.
Necrotizing enterocolitis (NEC) is a gastrointestinal disease that commonly affects preterm infants and is a major cause of morbidity and mortality in neonatal intensive care units (NICUs). Despite the advancements made in providing neonatal intensive care in recent years, NEC remains a devastating disease in NICUs. Approximately 7%–8% of premature infants in the NICU are diagnosed with NEC, with mortality rates approaching 20%–30% (1, 2). Of those that survive, many suffer from detrimental long-term effects on the intestines, growth, and neurodevelopment (3, 4).
NEC is characterized by inflammation and necrosis in the intestines, and often presents with a distended abdomen and blood in the stool (5, 6). Currently, NEC is treated with either a medical or surgical approach. The medical approach for the milder stages of NEC, consists of cessation of feedings, stomach decompression, antibiotics, frequent monitoring, and supportive care. Surgery is required if the infant experiences gangrene or intestinal perforation, and this treatment approach carries a higher rate of mortality (7). These treatment approaches have not changed in several decades and novel approaches to prevent or treat NEC are desperately needed.
Research into identifying the etiology of NEC has revealed that the most prominent risk factor is infant prematurity (8, 9). Approximately 9 of 10 infants diagnosed with NEC are born premature (gestational ages 22–37 weeks), with the most severe cases typically manifesting in very low birth weight (VLBW) preterm infants with a birth weight of <1,500 grams. Although cases of NEC have been observed in full-term infants, VLBW infants maintain the highest chances of contracting and succumbing to NEC (10).
This increased occurrence and fatality in premature infants has been attributed in part to their underdeveloped innate and adaptive immune systems, as well as decreased diversity of the gut microbiome compared to those of full-term infants (11, 12). Research suggests that intestinal immaturity and undeveloped immunity of preterm infants allows pathogens to bypass the epithelial cell layer to induce inflammation (13). One of the ways to decrease NEC incidence is to provide maternal breast milk to infants. Human milk oligosaccharides (HMOs) and immunoglobins (Ig), such as immunoglobulin A (IgA), are present in breast milk and have been shown to protect against NEC (14, 15). The components in breast milk help prevent the onset of NEC and shift the infant's gut microbial composition, which in turn bolsters the immune response (16). While we have some idea of the factors that contribute to and the factors that protect against the disease, the specific mechanisms that lead to the pathogenesis of NEC are not fully understood.
In this review, we examine factors that may contribute to NEC and associated pathogenesis, including the role that the mucosal immune response and the microbiome play in disease. Furthermore, we outline the various in vitro and in vivo NEC models used to demonstrate these findings and explore how these conclusions can lead to the development of preventative measures and treatments designed for NEC.
Although the etiology of NEC has yet to be completely elucidated, there are a multitude of factors, before and after birth, that can predispose infants to NEC. Maternal health status can provide substantial insight into an infant's risk of contracting NEC. According to a review of NEC risk factors in infants, variables such as maternal age, intrapartum antibiotics, incomplete steroid exposure, and maternal high neutrophil to lymphocyte ratio (NLR) are significant prognostic factors (9). Several observational studies have examined these factors in detail. A retrospective case control study with 97 matched pairs of infants showed a significantly higher odds ratio for antenatal ampicillin exposure for infants who later developed NEC than control infants (17).
Considering antenatal steroid exposure, it has been established that this treatment reduces morbidities and improves overall neonatal survival. However, an incomplete course of antenatal steroids or no steroid exposure has been associated with higher rates of more severe NEC (18). In a separate retrospective cohort study, an elevated maternal NLR (indicative of systemic inflammation) was significantly associated with the development of NEC (19). It is critical to note that blood NLR is a key diagnostic and prognostic indicator for disease states such as diabetes, obesity, hypertension, and heart disease, which are marked by inflammation. As such, the positive association between elevated maternal NLR and NEC suggests a possible relationship between NEC and placental vascular dysfunction caused by these disease states.
Preeclampsia, a serious complication of pregnancy, is also associated with an increased risk of NEC in preterm infants. Although the pathogenesis of preeclampsia remains unclear, it is theorized that the placental ischemia, abnormal hemostasis, leukocyte activation, and dysregulated nitric oxide metabolism associated with preeclampsia seem to be core components that may contribute to NEC development in preterm infants (20). Overall, preeclampsia reduces placental perfusion, which can lead to fetoplacental hypoxia and the pathogenesis of intrauterine growth restriction (IUGR). Both IUGR and reduced placental support, as indicated by abnormal patterns in antenatal umbilical dopplers, can impose increased risks for later NEC development (20, 21). Additionally, maternal diabetes mellitus (DM) poses a significant risk of NEC to infants, as determined by a retrospective study of low birthweight infants born to mothers with and without DM (21, 22).
Birth route may also provide insight into an infant's risk of developing NEC due to the impact that birth route has on the infant microbiome. However, the effects of Cesarean section (C-section) on the risk of NEC development are highly contested. A recent retrospective review discovered that delivery by C-section (and the presence of an umbilical arterial catheter) is associated with a decreased risk of NEC, possibly due to a decreased stress burden on the neonate during the C-section birthing process as compared to vaginal birth (23). A secondary analysis of data from a randomized controlled trial found no significant association between C-section in extreme preterm delivery and the onset of NEC (24). In contrast, another national case-control study established a positive association between C-section and the risk of NEC (25). Thus, there is conflicting data describing the relationship between C-sections and NEC incidence in neonates. Such disparities in data further indicate that NEC is a multifactorial condition and additional studies are required to delineate the maternal conditions that may predispose an infant to the disease.
Infant prematurity, characterized by both low birth weight and gestational age, is one of the most important risk factors for the development of NEC. Several studies have established that infants with a lower gestational age have a greater risk of developing NEC, along with higher mortality and surgical need (26, 27). Another retrospective study reported a higher NEC incidence in preterm infants that are small for gestational age (SGA) (28). While NEC pathogenesis in SGA neonates has not been completely explained, it has been proposed that gastrointestinal (GI) tract ischemia can contribute to NEC pathogenesis in preterm infants. Immature development of the GI tract can prime a “leaky” gut barrier susceptible to bacterial translocation due to incomplete formation of tight junctions, impaired peristalsis, and a thin mucus layer (29). The reduced structural integrity of the gut barrier can further decrease the uptake of essential nutrients for growth, exacerbating the effects of NEC.
Different types of infant nutrition can impact the pathogenesis of NEC. The nutritional requirements of preterm infants usually cannot be sustained solely with breast milk or standard formula—bovine and human-milk-based fortifiers are often needed to provide additional proteins, fats, vitamins, and minerals for adequate growth and development. However, some studies suggest that bovine milk-based infant formulas are positively associated with a higher risk of NEC, reviewed in (30). Although the exact link between bovine milk-based standardized formulas and NEC pathogenesis is not clear, one theory suggests that in the absence of the protective factors found in breast milk, infants receiving formula are at an increased incidence of NEC. This may render the gut more susceptible to the overgrowth of pathogenic microbes, such as the family of Gram-negative Enterobacteriaceae, and the initiation of widespread pro-inflammatory responses to bacterial translocation across the gut barrier (31). In contrast, the administration of maternal breast milk has been conclusively established to decrease NEC incidence (32). It has been long-established that human milk is the ideal source of nutrition for both premature and full-term infants. Several studies have demonstrated that there is a clear benefit to maternal human milk or donor human milk in the absence of maternal milk, reviewed in (33). Premature infants who received human milk have a demonstrably lower incidence of NEC than those who did not (34).
Intestinal dysbiosis, or the imbalance of a healthy gut microbial composition, has also been implicated as a predisposing factor to NEC. It is known that the gut microbiome of preterm infants has considerably reduced bacterial diversity and increased vulnerability to pathogens as compared to full-term infants (35). Additionally, there is a positive association between early antibiotic use and NEC onset, which supports the intestinal dysbiosis hypothesis (36). There have also been reports of immune dysregulation in conjunction with intestinal dysbiosis, particularly concerning heightened toll-like receptor 4 (TLR4) signaling and downstream inflammatory responses (37). Taken together, the pathogenesis of NEC is multifactorial and complex, rendering the root pathophysiology of NEC largely unanswered.
Immature intestinal immune defense is among several factors associated with the high morbidity and mortality rates of NEC. Alteration of key innate and adaptive immune responses leads to dysfunction in intestinal barrier thus resulting in an increased inflammatory response (Figure 1) (38–40). The onset of NEC has been linked to low birth weight and gestational age, so while all infants have immature innate immunity, premature infants are also born with undeveloped adaptive immune systems. To make up for this weakened immunity, the transfer of maternal milk components, including secretory IgA (sIgA), as well as placental immunoglobulin G (IgG), provide protection to newborns until their own adaptive immune defenses can develop (15). In formula-fed premature infants, the levels of transferred maternal immune defenses are significantly reduced, potentially increasing their susceptibility of developing NEC (41).
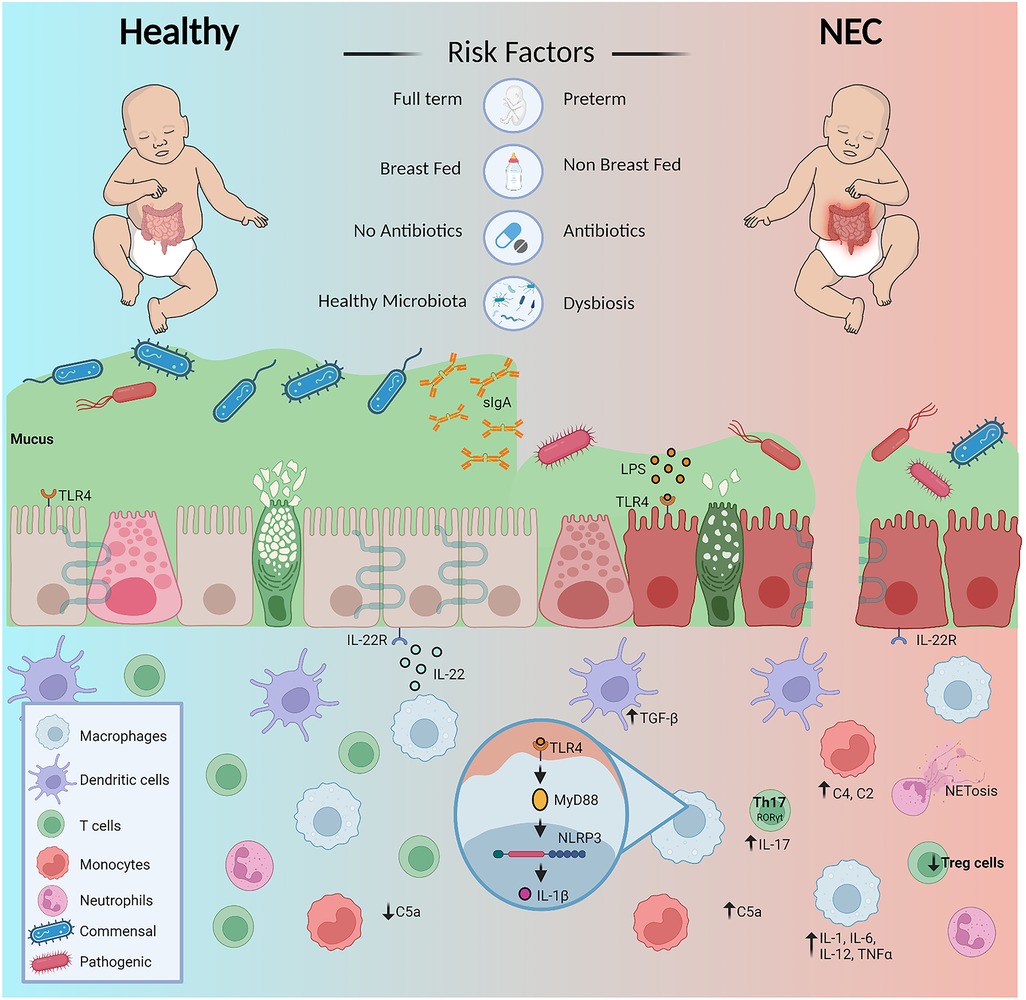
Figure 1. Diagrammatic overview of factors predispose premature infants to NEC and dysregulation of immunity contributing to the diseased state. Figure created with Biorender.com and affinity designer.
In this section, we summarize the current scientific evidence of the innate and adaptive immune responses in infants with NEC. Specifically, we discuss how NEC pathogenesis relates to the vertical transfer of immunity from mother to child, alteration in physical barriers, and immunity guarding the gastrointestinal tract.
Newborns do not cultivate a fully mature immune system until a few years after birth (42). To compensate, maternal IgG and IgA antibodies are donated from the placenta and maternal breast milk (if provided) to protect against pathogens and the development of NEC (15). Maternal IgG transfer to the fetus across the syncytiotrophoblast depends on the IgG-FcRn (neonatal Fc receptor) interaction. The expression of IgG-FcRn begins during the first trimester (12 weeks) of pregnancy and continues to rise until between 17 and 41 weeks gestation. The majority of placental IgG transfer occurs after 28 weeks of gestation. IgG levels reach 50% maternal concentration between 28 and 33 weeks gestation and will rise above maternal levels by 20%–30% at term (43, 44). It is possible that low IgG levels in preterm infants may predispose these infants to develop NEC.
In addition to the transfer of maternal IgG, transfer of maternal IgA through breast milk, also protects infants from NEC. Originating from IgA+ plasma cells in the gut and educated by gut microbiota, IgA in the intestine can bind to pathogens and aid in their clearance. The ability of bacteria to bind to IgA was negatively correlated to NEC development, and the reduced stool bacterial diversity known to precede NEC was associated with a higher amount of unbound Enterobacteriaceae (15). Taken together, this data suggests that the absence of sIgA creates higher susceptibility to infections as well as delayed gut microbiota maturation which leads to gastrointestinal inflammatory diseases such as NEC.
As the primary source of nutrition, breast milk ensures proper growth and development for newborns. Human milk is composed of micro and macronutrients, bioactive components, growth factors, antibodies, and HMOs (45). HMOs, in particular, play an important role in shaping microbiome composition and modulating neonatal immunity. HMOs act as natural prebiotics, functioning as soluble decoy receptors or antiadhesives to block the adhesion of pathogens to epithelium. They also enhance commensal growth and limit pathogen growth (46). HMOs are non-digestible sugars, composed of five basic monosaccharide units: glucose, fucose, d-galactose, N-acetylglucosamine, and sialic acid (47, 48). These monosaccharide units are joined by glycosidic linkage to generate a variety of HMOs with different functions. HMOs are indigestible in the human upper digestive tract and remain intact while in the colon. Colonic microbes secrete enzymes to utilize these HMOs as nutrition (49, 50). Many of the commensals that degrade HMOs for fuel are members of the Bifidobacterium genus, mostly beneficial bacteria for infant health. Specific examples are B. longum and B. breve that are usually prominent in the digestive tract of breastfed infants.
In addition, Bacteroides species possess an excellent capacity to metabolize dietary polysaccharides to host-derived mucus-associated glycans. A study by Sodhi and colleagues has shown that HMOs 2′-fucosyllactose (2′-FL) and 6′- sialyllactose (6′- SL) can reduce NEC severity through TLR4 inhibition (51). 2′-FL also suppresses lipopolysaccharide (LPS) induced inflammation during Escherichia coli (E. coli) invasion of intestinal epithelial cells (52). Similarly, Masi et al. found significantly lower disialyllacto-N-tetraose (DSLNT) in the maternal milk given to infants prior to NEC development (53). Furthermore, authors reported that low DSLNT in milk was also associated with a significantly lower relative abundance of Bifidobacterium sp. and higher Enterobacter cloacae in the stool of infants prior to NEC (53). Fractions of HMOs were also shown to decrease mucus penetrability and bacterial attachment by enhancing the expression of Mucin 2 (MUC2) in a mouse model of NEC (54).
Other milk factors such as casein, a highly glycosylated breast milk protein, promotes intestinal defenses by increasing goblet cell numbers, enhancing Muc2 expression, and Paneth cell activity (55, 56). Additional factors found in breast milk include lactoferrin and lysozymes that possess antipathogenic properties. Enteral supplementation of lactoferrin has been shown to decrease the likelihood of late-onset bacterial and fungal sepsis in preterm infants, but meta-analysis has shown there was no significant decrease in NEC in infants who were received lactoferrin (16). Breastmilk platelet activating factor-acetyl hydrolase (PAF-AH) potentially protects preterm newborns from NEC (57). Similarly, interleukin-10 (IL-10) found in breast milk has been found protective against developing NEC in premature infants (58). In addition to IL-10, maternal transforming growth factor beta (TGF-β) provides protection by helping to increase IgA locally in the gut (59). Growth factors found in breast milk, such as insulin-like growth factor (IGF) and epidermal growth factor (EGF), support intestinal health and may protect against the development of NEC (60–65).
Mucus is one of the first lines of intestinal host defense. Mucus is produced by goblet cells, which are found in the crypts of Lieberkühn. The colonic mucus layer is divided into two layers, an outer, penetrable layer, and an inner, impenetrable layer. This contrasts with the mucus in the small intestine (SI) which is single layered and penetrable by bacteria. A protective layer of mucus keeps bacteria in the SI away from the intestinal epithelium by antimicrobial proteins (AMPs) secreted by Paneth cells (66). Studies have found defective and a significantly lower number of goblet and Paneth cells in the SI of infants with NEC compared to NEC (67). Using HT29-MTX-E12, a mucus secreting cell line, Hall and colleagues reported that breast milk significantly lowered the adherence and internalization of NEC-associated pathogenic E. coli into the mucus compared to infant formula, suggesting that breast milk enhances mucus integrity (68). Clostridium difficile (C. difficile), a known gut pathogen, also influences mucus production and composition (69).
Antimicrobial peptides (AMPs), such as defensins, including human β-defensin-3 (hBD3), cathelicidins, C-type lectin receptors (CLRs), regenerating islet-derived protein 3, and intestinal enzymes such as phospholipase A2-IIA (PLA2) and lysozyme are expressed in the gut epithelium and provide protection for the intestinal mucosa from pathogenic bacteria either by killing pathogens or by inhibiting their growth (70, 71). In addition, AMPs are involved in the immune response and shaping the microbiome (72). Using an experimental rat NEC model, Underwood and colleagues found increased intestinal mRNA expression of the AMPs lysozyme, secretory PLA2, and pancreatic-associated proteins 1 and 3 in rats with NEC compared to either dam-fed or formula-fed rats supplemented with the probiotic bacteria Bifidobacterium bifidum (B. bifidum), suggesting that AMP induction is a mucosal response to gut inflammation in NEC (73). Another study evaluated the defensin hBD3, a small cationic antimicrobial peptide that can exert multiple protective properties on the gut. Using an animal NEC model, Sheng et al., showed that hBD3 administration decreased the incidence of NEC, reduced NEC severity (decreased pro-inflammatory cytokines, intact intestinal barrier), and increased the survival rate of the animals (74). Collectively, these studies suggest a protective role for mucus and associated AMPs in neonatal mucosal defense and intestinal barrier function in NEC.
During infection, complement proteins assist in the phagocytosis of invading pathogens by opsonization, generating inflammatory responses, and altering the activity of B and T lymphocytes (75, 76). Three different pathways—lectin, alternative, and classical—activate the complement cascade. Previous studies have reported defective complement protein activity in preterm infants (77, 78). More specifically, one study reported low complement component 3 (C3) and complement component 9 (C9), intermediates of complement pathways, in preterm infants (79, 80). C5a, a cleavage product of complement component 5 (C5), is a potent chemoattractant, anaphylatoxin, and intermediary in both the conventional and non-canonical complement pathways. C5a was reported to be substantially expressed in NEC cases and could be partially responsible for inflammation in NEC. Due to its multifaceted nature, C5a is being studied for its utility as a clinical marker for the diagnosis of neonates with NEC in conjunction with radiographic evidence of disease (81). In addition, MBL-associated serine protease-2 (MASP-2), an enzyme associated with C2 and C4 cleavage and activity, is detected in higher concentrations in the cord blood of premature children who are susceptible to NEC and is linked to a threefold increased risk of developing NEC (82, 83).
Drosophila Toll was discovered as a receptor for dorso-ventral patterning during development and was later identified as a participant in immunity against fungal infections (84). Consequently, several other homologues of Toll, named Toll-like receptors (TLRs) were discovered in mammals. TLRs sense pathogen-associated molecular pattern molecules (PAMPs) and danger-associated molecular patterns (DAMPs) through their N-terminal extracellular leucine-rich repeats (LRRs) and elicit innate immunological responses, including the production and release of inflammatory cytokines (85). So far, 10 different types of TLRs have been identified in humans and 12 in mice. TLR1, TLR5, TLR6, and TLR10 are membrane receptors that may detect extracellular ligands while TLR3, TLR7, TLR8, and TLR9 work on subcellular structures. For example, TLR9 is found on endosomes and recognizes nucleic acids derived from pathogens and self-damaged cells (85, 86). TLR2 and TLR4 are expressed on the cell membrane as well as on subcellular structures.
TLR4 is a receptor for LPS, a component of Gram-negative bacteria's outer membrane that is critical for the NEC pathogenesis (87). TLR9 binds to and is activated by unmethylated cytosine-guanine oligodeoxynucleotides (CpG ODNs) in bacterial genomes, and acts as antagonist of TLR4. Activation of TLR4 in newborn mouse epithelial cells by LPS results in undesired activation of the NF-κB pathway that leads to damage of the intestinal mucosa through production of pro-inflammatory cytokines, which is one of the hallmarks of NEC (87). Several studies have shed light on the association of TLR4 with NEC (41). Recently, Liu and coworkers have shown both TLR4 and necro apoptotic protein upregulation in both NEC patients with NEC and animal NEC models (88). Egan et al., highlighted the role of TLR4 in recruiting the inflammatory CD4+ Th17 cells into the intestinal mucosa via activation of cognate chemokine ligand 25 (CCL25) in NEC (89). In an another study, Colliou et al., found a commensal Propionibacterium bacterial strain named UF1 that can reduce intestinal inflammation through the reduction of Th17 cell expansion in the gut of a mouse NEC model (90). TLR4 activation significantly inhibits the β-catenin signaling that is important for enterocyte proliferation in the ileum of newborn mice, which further leads to apoptosis and can lead to NEC (91). Studies have shown that activation of TLR9 can decrease experimental NEC severity, and that TLR9 activation can inhibit TLR4 signaling via IL-1R-associated kinase M (92, 93). In addition to TLR9, NOD2 reduces NEC severity via suppressing TLR4 and genetic variants in NOD2 are associated with NEC development (94, 95).
Originating from myeloid cell lineage monocytes, macrophages (Mϕ) act as a frontline guard of innate immunity against invading pathogens. Monocytes and Mϕ have several weapons in their arsenal to tackle incoming threats. By recognizing molecular patterns via toll-like receptors (TLRs), nucleotide-binding oligomerization domain-containing proteins (NOD2), and C-type lectin receptors (CLRs,) these cells either actively engage in phagocytosis or secrete various cytokines and chemokines to alert and recruit other immune cells (96). Classical monocytes (CD14+CD16−), intermediate monocytes (CD14+CD16+), and non-classical monocytes (CD14dimCD16+) are the three subsets of human monocytes. In mice, monocytes are grouped based on expression levels of lymphocyte antigen 6 complex (Ly6C+ and Ly6C−) on their cell surface (97).
Several studies have suggested that tissue infiltration and enrichment of monocyte-derived Mϕ occur during inflammation in NEC (98–100). Intestine monocyte-derived Mϕ are nonproliferative, short lived and terminally differentiated, rendering their continuous replacement necessary for homeostasis. A study by Managlia et al., revealed the significance of nuclear factor kappa B (NF-κB)-driven monocyte activation, recruitment, and differentiation in neonatal intestines in NEC (99). They concluded that NF-κB-mediated activation and differentiation of Ly6c+ monocytes into Mϕ and their recruitment into the intestine are critical for NEC development and disease progression. Olaloye and colleagues have identified a novel subtype of inflammatory CD16+CD163+ monocytes/Mϕ associated with infants with NEC (100). In infants with NEC, peripheral monocyte counts drop due to their recruitment to the damaged intestine (101). Following recruitment, monocytes undergo differentiation to form pro-inflammatory M1-type Mϕ (102). Monocyte-derived M1 Mϕ in humans and in animal models have been linked to the severity of NEC (102, 103). Interferon regulatory factor 5 (IRF5), a factor crucial for M1 Mϕ polarization is highly expressed in infants with NEC compared to controls. Specifically, IRF5 deficiency significantly reduced M1 polarization, inflammation, and intestinal injury in experimental NEC (103). Inflammation and intestinal cell damage caused by M1 Mϕ is linked with their high level of pro-inflammatory cytokines such as IL-1, IL-6, IL-12, chemokines (Ccl4, Ccl5), and tumor necrosis factor (TNF) production. By inhibiting M1 and promoting M2 polarization of Mϕ, heparin-binding epidermal growth factor (HB-EGF) has also been found to protect against experimental NEC (102).
As one of the most abundant immune cells (nearly 70% of total leukocytes) in human blood, neutrophils are among the first responders in the fight against potential pathogens or tissue damage/injury. Neutrophils eliminate pathogens either by recruiting a wide variety of immune cells through the secretion of cytokines, chemokines, and leukotrienes or by causing direct damage to tissue or pathogens by releasing lytic proteases and neutrophil extra cellular trap (NETs) (104). In addition to their well-documented protective role, neutrophils are also able to cause significant tissue damage through the release of reactive oxygen species (ROS) in intestinal inflammation (105).
Early neutropenia has been associated with higher odds of developing NEC (106). Interestingly, neutrophils in preterm newborns have altered immunological functions, including impaired phagocytosis. Another study by Zindl and colleagues revealed the protective role of IL-22-producing neutrophils in experimental colitis by increasing AMP production and promoting mucosal repair (107). In the context of NEC, a recent study from Mihi et al., demonstrated a protective role of IL-22 treatment in attenuating intestinal injury and enhancing epithelial proliferation in experimental NEC (108). This study also found that there was a lack of IL-22 production in preterm infants or developing mice, suggesting that immunomodulatory treatments may help protect premature infants from the intestinal inflammation seen in NEC.
As specialized antigen presenting cells (APCs), dendritic cells (DCs) serve as critical link between innate and adaptive immunity. In intestine, DCs are present in Peyer's patches, mesenteric lymph nodes (MLNs), and the colonic lamina propria to provide protection against invading pathogens. To date, several studies have highlighted the protective role of DCs in regulating the gut inflammation; however, studies investigating the role of DCs in NEC is limited. In one study, which utilizes Cronobacter sakazakii (C. sakazakii) to induce NEC in mice, Emami and colleagues have reported higher DC recruitment in mouse gut. They found that DC recruitment to the gut accelerated the destruction of the intestinal epithelium and promoted NEC onset with increased TGF-β production (109). C. sakazakii also induced pyroptosis in the intestinal epithelium and promoted NEC by induction of IL-1β and Gasdermin D (GSDMD) through TLR4/MyD88 mediated activation of the nucleotide-binding oligomerization domain (NLRP3) inflammasome (110). Another study by Nolan and colleagues investigated the role of aryl hydrocarbon receptor (AhR) signaling in DCs during experimental NEC, as this signaling pathway helps regulate intestinal immunity and homeostasis. They found that a lack of AhR signaling in DCs increased NEC-mediated intestinal inflammation, and that this effect was associated with an increase in a specific subset of macrophages in the small intestinal lamina propria (111).
Adult human intestine is made of a single layer of epithelium, covering an area of 32 m2 (112). The intestinal epithelium is important for digesting food and absorbing nutrients, but it is also the largest entry port for pathogens. To provide protection against these pathogens, “as a guard of port”, complex and tightly controlled innate and acquired immunity are required. Among the many different types of immune cells involved in this protection are intraepithelial lymphocytes (IELs). IELs are positioned between intestinal epithelial cells and constantly patrol the epithelial barrier (113). IEL subsets, composed of antigen-experienced T cells, are in direct contact with enterocytes and antigens in the gut lumen. These cells are classified based on the expression of T cell receptor-γδ (TCRγδ)+ and TCRαβ+ (114). Approximately 60% of small intestinal IELs are TCR+ cells. γδ IEL play a crucial role in mucosal defense by regulating the production of IgA, clearing and repairing damaged epithelium, increasing production of TGF-β cytokines and by decreasing IFN-γ and TNF-α in response to stress and infection (115). The protective role of IELs is also evident in TCRγδ-deficient mice, as these mice have defective gut epithelial morphology and impaired IgA production (116). When compared to non-NEC controls, surgical NEC patients with NEC had a lower number of γδ IELs in the ileum (116). Researchers have shown that subsets of IELs are dependent on AhR activation for their survival (117). However, a recent study did not find any involvement of IELs in AhR activation-mediated protection against NEC, indicating that the protective role of IELs against NEC is not AhR-mediated (118).
In addition to IELs, infants with NEC also have altered functions of some subsets of CD4+ T cells, Th17, and regulatory T (Treg) cells (89, 119–121). Th17 cells are strongly implicated in intestinal inflammation and are linked with the pathogenesis of NEC. In infants with NEC, Pang and colleagues found a lower percentage of Foxp3-expressing Tregs with several functional defects, including the inability to block IL-17 expression (121). In NEC tissue, Th17 cells appear to cause intestinal damage that is reduced by IL-17 receptor inhibition by STAT3 activation (122). Additionally, retinoic acid-induced polarization of CD4+ T cells towards Treg from Th17 resulted in reduced NEC severity (123). Furthermore, Zhao et al. reported an increased percentage of RORγt+ cells (inflammatory Th17 and type 3 innate lymphoid cell populations) in the intestinal lamina of mice and humans with NEC compared to those without NEC (84). Studies have also demonstrated a significant decrease in lamina propria associated Treg cells in surgical NEC specimens (85, 86, 89). In addition, a Treg/Th17 imbalance leads to the excessive proinflammatory response preceding tissue injury and necrosis associated with NEC development (122).
Although the direct association between the microbiota and the pathogenesis of NEC is not well understood, mounting evidence suggests a link between early gut microbiota dysbiosis and NEC (87, 88, 90). Probiotic supplementation to premature neonates has been shown in some studies to decrease the incidence or severity of NEC, further establishing the relationship between NEC and microbiota (91–94).
Early microbiota composition and its diversity in the gut of newborn infants is mainly influenced by delivery mode, antibiotic exposure, human milk feeding, and time spent in the NICU. Vaginally born infants not only develop stronger immunity but also are predominantly colonized by beneficial microbes such as Lactobacillus sp. present in mother's vaginal microbiota (95). Members of Lactobacillus are well known to prevent pathogen colonization by lowering the pH or by secreting inhibitory compounds (124, 125). The microbiota of infants born via C-section resemble the mothers' skin microbiota in early life and lack members of Bacteroides species that are present mostly in vaginally-delivered infants (126).
In addition to delivery mode, feeding also affects microbiome composition and diversity. Formula-fed newborns have lower overall bacterial diversity, lesser beneficial bacterial number, and a higher number of pathogenic bacteria like Clostridium sp. compared to breast-fed infants (127). Clostridium sp. and their secreted toxins can be associated with NEC severity (128, 129). Time spent in the NICU with lifesaving machines attached to preterm infants including, ventilators, and incubators, have also been shown to harbor pathogenic bacteria including members of Streptococcus, Klebsiella, Staphylococcus, Neisseria, and Enterobacteriaceae communities (130–133). Members of the phyla Firmicutes, such as coagulase-negative staphylococci (CoNS) and Proteobacteria are implicated in NEC pathogenesis, however, many of their members are also found in healthy infants (134). Higher bacterial relative abundance from the class Gammaproteobacteria, namely C. sakazakii, Klebsiella sp., E. coli, and those from the phylum Proteobacteria are also present in the feces of infants who develop NEC (135). In addition to bacteria, viral presence is also associated with NEC. Stool analysis from 51 infants with NEC and 39 controls demonstrated that the presence of adenovirus and Epstein-Barr virus are associated with NEC severity (136). In another recent study, stool samples obtained from 9 infants with NEC infants and 14 controls matched for weight and gestational age, showed reduced viral beta diversity over the 10 days before NEC onset. This study also identified that viral NEC-associated contigs belonging to Myoviridae, Podoviridae and Siphoviridae are associated with the time period 0–10 d post NEC onset (137).
With the high prevalence of NEC, the need for effective in vivo models has become more important in recent years. Due to the aggressive nature of the disease and the scarcity of available human specimens, performing experiments with human samples is difficult and multi-center studies are typically needed (138). As a result, animal models are commonly used to study NEC by inducing inflammation that mimics the intestinal damage seen in human infants.
While the conditions of in vivo experimental NEC models are generally based on similar underlying principles, several different animals have been used to study NEC (Figure 2). The rat's intestinal development is similar to a human premature infant, making it an excellent model for investigating preventative measures and therapeutics for NEC (139). Early studies using a rat model concluded that the gut microbiota and the absence of breast milk are significant factors in NEC pathogenesis (140, 141). Further, several laboratories have used hypoxia, LPS, and hypothermia at different time points in a day for several days to help induce NEC in laboratory settings (142). Due to their affordability, preterm survivability, and resistance to typical stressors used to develop the disease, rat models are a desirable option when investigating NEC but rats are not ideal for research at the genomic level. Their slower development and challenges with culturing embryonic stem cells in rats makes it difficult to generate transgenic lines compared to mice (143, 144). These shortcomings necessitated the creation of other types of animal NEC models.
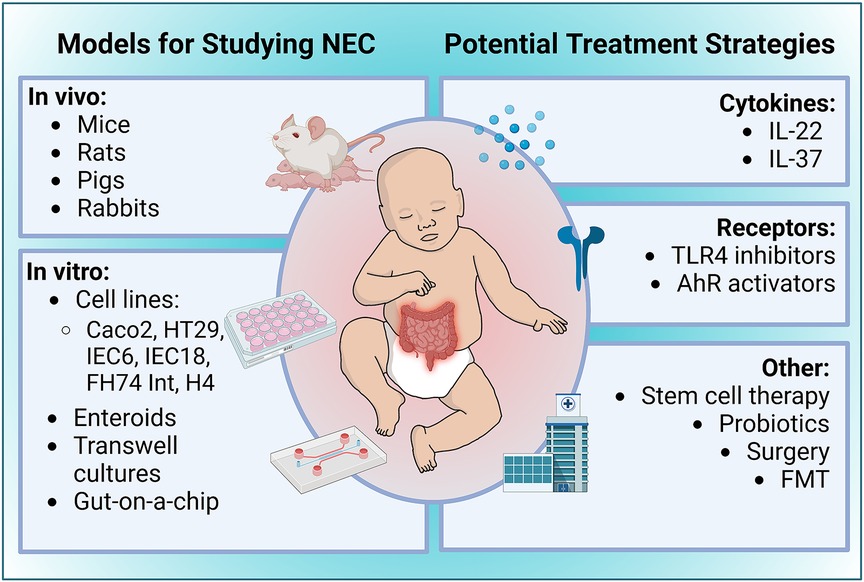
Figure 2. Overview of experimental models of NEC and potential treatment strategies. Figure created with Biorender.com.
Although their small size makes them technically challenging to work with, mice are the preferred model for genomic studies as it is far easier to create transgenic colonies. Another appealing feature of the mouse model is its experimental flexibility, with some models successfully inducing NEC by beginning the gavage feed at postnatal day 4 while others begin at postnatal day 7 (145, 146). However, mice delivered more than one day prior to the determined due date have a 100% mortality rate (147). Because of this low viability, it is extremely difficult to use a preterm mouse model for studies that require animals to be delivered via cesarean section.
Pigs share many features of anatomy and physiology with humans, rendering them one of the more popular choices when exploring NEC pathogenesis. Additionally, the piglet's larger size affords the ability to study preterm neonates (148, 149). Piglets are a good model for testing preclinical drugs, effects of various diet formulations, and pathological manifestation on NEC (150). While it is true that hypoxia and hypothermic stress induces histological changes that resembled NEC in piglet models, the inflammation induced by this model is not always contained within the lower gastrointestinal tract, with some instances reported of inflammation spreading to the stomach and jejunum (139, 150–152).
Rabbit NEC models are infrequently used but have been used to study the effects of NEC that extend past the gut. Non-human primate models, although rare and expensive, have also been used as an experimental NEC models due to the homology to humans in both anatomy and at the genomic level (139).
In vivo animal models allow for limited NEC modeling as the cellular genetics, drug metabolism, immunology, gut microbiomes, and HMOs can differ significantly from humans. In vitro intestinal models used to study NEC are briefly summarized in this section and have been covered extensively elsewhere (153–156).
Different in vitro models such as the human epithelial cell line Caco2, colon adenocarcinoma derived cell line HT-29, IEC-6 and IEC-18 derived from rat SI, and most importantly, fetus derived FHs 74-Int and H4 cells are frequently used in in vitro NEC studies (153). These cell lines are optimized and phenotypically mimic different regions of the gut including ileum, duodenum, jejunum, and colon, each requiring specific culturing conditions.
Recent scientific advancements in culturing human intestinal organoids (enteroids) also called “mini guts”, allow investigators to recapitulate the intestinal cell morphology that is crucial for studying the molecular mechanisms of NEC. Enteroids derived from LGR5+ progenitor cells of the SI and colon, allow for the study of barrier function, gut inflammation, cell proliferation, drug responses, and intestinal microbial interactions characteristic of NEC (157). Further advancements of in vitro models led to the development of a “gut-on-a-chip”, a method which cultures intestinal cells to mimic the microenvironment of the intestine (158, 159). The gut-on-a-chip model provides a suitable environment to culture different human cell types including epithelial, endothelial, and immune cells with gut microbes together in a controlled environment, to explore gut physiology and inflammatory changes seen in NEC, and can also be used as a pharmacological platform to test potential drug treatments (160).
Though, these in vitro models excellently resemble human intestine, several key criteria are considered in cell culture model design. Table 1 compares common different models and devices, specifically summarizing whether the models are static or microfluidic, in vitro or ex vivo, cell differentiation, cell polarity (apical out or basal out), nutrient absorption, drug metabolism, crypt villus formation, mechanical stimulation or peristalsis, oxygen gradient modulation, measure trans epithelial electrical resistance (TEER), co-culture with endothelial, vascular, and immune cells, and co-culture with gut microbes.
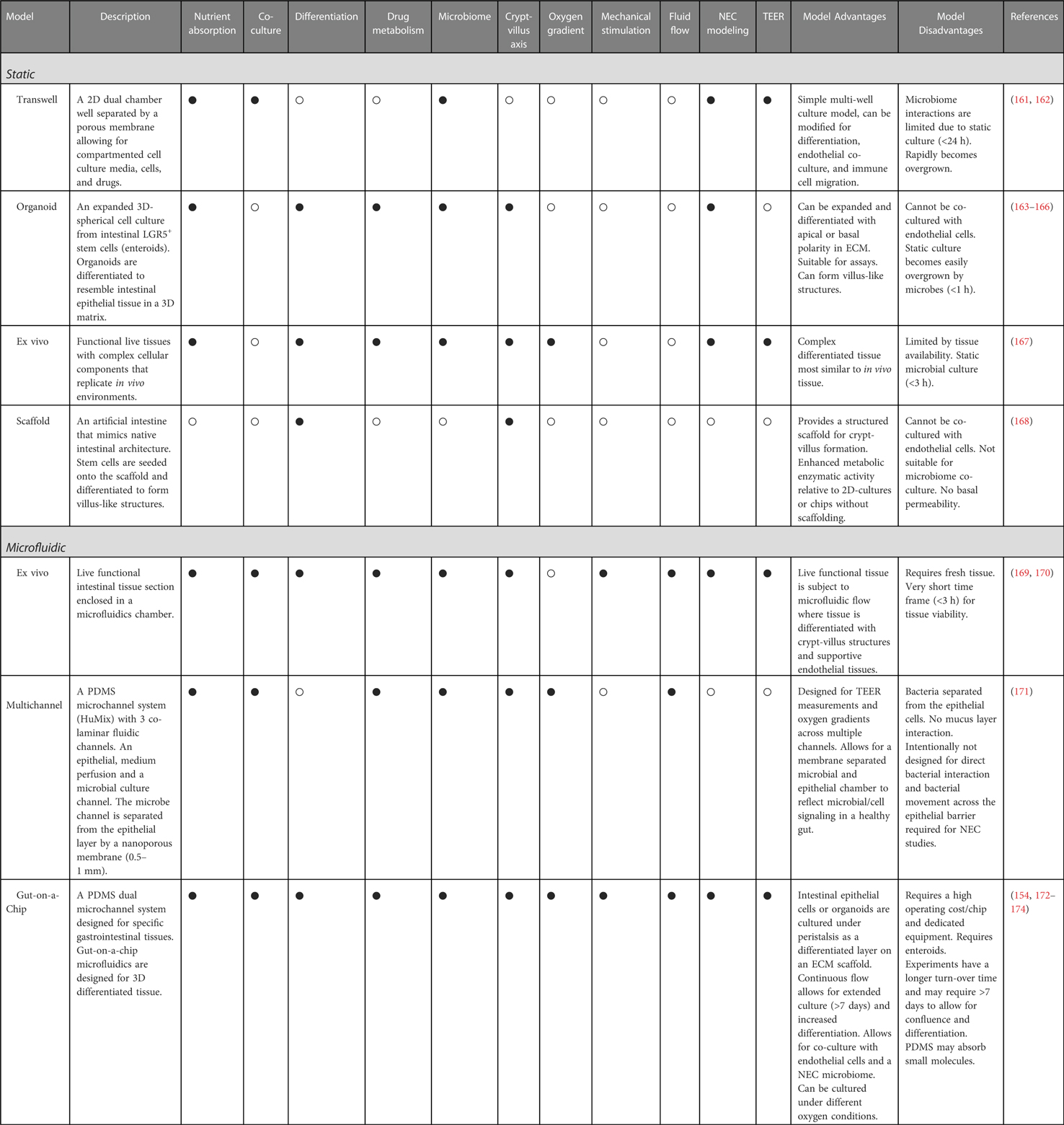
Table 1. Characteristics and limitations of in vitro static and microfluidic devices for NEC disease modeling (• = yes, ο = no).
Static models are standard tissue culture models which include “NEC-on-a-dish” 2D, 3D organoid and transwell culture models (175). Additionally, synthetic scaffolds, and ex vivo tissue (Ussing chambers) are used to measure live tissue (167, 168). Static models use growth factors to differentiate intestinal epithelial cells (IECs) and organoids, derived from LGR5+ progenitor cells, into diverse functional intestinal cells (163). Static models are generally less time consuming, less expensive, and more accessible, but are relatively limited to the degree of differentiation, co-culture, and microbiome interactions. Typically, in static models, microbiome interactions are limited to between 1 and 24 h based on the model due to rapid microbial overgrowth in static conditions.
Gut-on-a-chip microfluidic devices use soft lithography to layer polydimethylsiloxane (PDMS) or micromilling to produce luminal and vascular channels separated by a porous membrane (reviewed in (176). Short term ex vivo microfluidic devices can evaluate live tissue conditions under constant flow (169, 170). The luminal flow in a microfluidic model enhances differentiation and 3D villus and crypt-villus like topography where adjacent air channels are regulated to mimic peristalsis through mechanical stimulation, thus providing a major advantage over static models. The NEC microbiome and HMO interactions, drug metabolism, and tissue integrity assays can be measured within the microfluidic chip system (177, 178). A major advantage of the microfluidic flow is that it reduces the static overgrowth of microbes, in turn reducing the limitations on the microbial co-culturing time to more than 7 days, depending on the specifics of the model. Gut-on-a-chip models can additionally be cultured under oxygen gradient modulation. Intestinal disease pathology is increased by lower oxygen gradients which induce Hif1-α signaling (179). Oxygen gradients under aerobic, hypoxic, and anaerobic culturing conditions have also been applied to resemble microbial intestinal environments under inflammatory conditions (176).
The several known risk factors of NEC discussed in this review provide promising treatment targets for NEC (Figure 2). One such treatment is IL-22, a cytokine belonging to the IL-10 family that is involved in epithelial cell regeneration, maintenance of gut barrier integrity, and tempering intestinal inflammation by mediating the microbiome (180). Given the observations of the versatile roles that IL-22 plays in gastrointestinal physiological processes and pathologies, especially as a stabilizer of intestinal homeostasis, there is a strong foundation to investigate the role of IL-22 in the context of NEC pathogenesis. As mentioned above, a recent study by Mihi et al., showed that neonatal mice and humans lack intestinal IL-22 production during NEC and supplemental administration of IL-22 attenuated experimental NEC severity, decreased intestinal inflammation, and enhanced intestinal epithelial repair (108). Additionally, IL-22 administration induced the expression of antimicrobial genes such as Reg3γ and fucosyltransferase 2 (Fut2). The AMP Reg3γ has been shown to protect the intestinal mucosa against pathogenic infections by limiting their expansions. Given this protective role of IL-22 in the experimental murine model of NEC, it is imperative that IL-22 administration be further investigated as a therapeutic for infants with NEC (108).
Another study by Cho et al., highlighted the importance of another cytokine, IL-37 in attenuating the inflammation in NEC (181). The study found that transgenic IL-37 pups were completely protected from inflammation caused by IL-1β, IL-6, TNF, and IL-17F compared to wild-type mice. In addition, IL-37 treatment restored the expression of cytokines Il4, Il13, and Il33 to baseline levels. Further, authors found that IL-37-mediated protection against NEC is largely achieved through modulation of the TLR repertoire (reducing TLR4 expression and inducing TLR5, TLR7, TLR9, and TLR13), and prevention of NEC-induced dysregulation of adaptive immunity (181).
Another promising treatment modality is the use of TLR4 inhibitors to mediate intestinal injury propagated by NEC. Hackam and colleagues have published several studies indicating that expression of TLR4 and members of its gene family render the premature intestine more susceptible to inflammation. Therefore, exploring TLR4 modulation or inhibition as a model for NEC treatment may be valuable. Lien et al., and Tidswell et al., noted the synthetic inhibitor eritoran tetrasodium (E5564) bound well to TLR4 (182, 183). Based on the structure of this inhibitor, an in silico search and screening of small molecule libraries conducted by Hackam and colleagues pinpointed a family of TLR4 inhibitors that reduces intestinal inflammation in experimental NEC (184, 185). Particularly, the compound C17H27NO9 (C34), a 2-acetamidopyranoside, significantly reduced NEC incidence in animal models and decreased TLR4 signaling ex vivo in resected ileum from infants with NEC (185). Indeed, these findings indicate C34 and its analogs are lead compounds for TLR4 inhibition that can provide therapeutic value and improve clinical treatments for NEC. In a recent study Lu et al., showed that activation of AhR either by its ligand indole-3-carbinol or by breast milk components prevented experimental NEC through inhibition of TLR4 signaling (118).
Stem cell therapy is another treatment option currently being explored because of anti-inflammatory properties with a focus on bone marrow-derived mesenchymal stem cells (BM-MSCs). Several studies have demonstrated that BM-MSCs extracted from mice, rats, and humans significantly reduce both NEC incidence and severity (186–188).
Amniotic fluid-derived stem cells (AF-MSCs) have also been investigated as a potential source for NEC treatment. A study by Zani et al., established that intraperitoneal injections of AF-MSCs in a murine model are significantly associated with a reduction in the incidence and severity of NEC and improved gut barrier function (5). Subsequent confirmatory studies verified that AF-MSC injections decrease histologic injury in experimental NEC models (189). Thus, there is indication that AF-MSCs have considerable beneficial effects as an inflammatory modulator and should be examined further as a therapeutic for NEC.
Experimental results of supplementation with probiotics and potentially fecal microbiota transplant (FMT) has also shown promising outcomes to treat NEC, however, appropriate donor selection, screening of FMT material, and a dosing strategy still need to be standardized (190–192).
NEC is a common gastrointestinal disease in premature infants associated with high morbidity and mortality. In recent years, substantial progress has been made to delineate the molecular mechanisms underlying the pathogenesis of NEC. The holistic approaches with scientific advancement to understand the risk factors predisposing an infant to NEC, including maternal, genetic, nutritional, and immunological changes in infants, clearly hold the potential to improve and lead to development of preventative measures and treatments to combat NEC. Although translating fundamental experimental discoveries to the bedside in the NICU is substantially challenging, continuous scientific efforts and collaborations between those working “at the bench” making discoveries in laboratories with those clinicians “at the bedside” caring for infants with NEC can lead to ground-breaking discoveries and transform the management of this devastating disease.
DKS, CM, KAO, MD, SM and MG reviewed the relevant literature, drafted, revised, and approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
MG is supported by National Institutes of Health (NIH) grants R01DK124614, R01DK118568, and R01HD105301, and the University of North Carolina at Chapel Hill Department of Pediatrics.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Fitzgibbons SC, Ching Y, Yu D, Carpenter J, Kenny M, Weldon C, et al. Mortality of necrotizing enterocolitis expressed by birth weight categories. J Pediatr Surg. (2009) 44:1072–5; discussion 1075. doi: 10.1016/j.jpedsurg.2009.02.013
2. Battersby C, Santhalingam T, Costeloe K, Modi N. Incidence of neonatal necrotising enterocolitis in high-income countries: a systematic review. Arch Dis Child Fetal Neonatal Ed. (2018) 103:F182–9. doi: 10.1136/archdischild-2017-313880
3. Bazacliu C, Neu J. Necrotizing enterocolitis: long term complications. Curr Pediatr Rev. (2019) 15:115–24. doi: 10.2174/1573396315666190312093119
4. Garg PM, Paschal JL, Zhang M, Pippins M, Matthews A, Adams K, et al. Brain injury in preterm infants with surgical necrotizing enterocolitis: clinical and bowel pathological correlates. Pediatr Res. (2022) 91:1182–95. doi: 10.1038/s41390-021-01614-3
5. Zani A, Pierro A. Necrotizing enterocolitis: controversies and challenges. [version 1; peer review: 3 approved]. F1000Res. (2015) 4:F1000 Faculty Rev-1373. doi: 10.12688/f1000research.6888.1. eCollection 201526918125
6. Patel RM, Ferguson J, McElroy SJ, Khashu M, Caplan MS. Defining necrotizing enterocolitis: current difficulties and future opportunities. Pediatr Res. (2020) 88:10–5. doi: 10.1038/s41390-020-1074-4
7. Kosloske AM. Indications for operation in necrotizing enterocolitis revisited. J Pediatr Surg. (1994) 29:663–6. doi: 10.1016/0022-3468(94)90736-6
8. Stoll BJ, Hansen NI, Bell EF, Walsh MC, Carlo WA, Shankaran S, et al. Trends in care practices, morbidity, and mortality of extremely preterm neonates, 1993–2012. JAMA. (2015) 314:1039–51. doi: 10.1001/jama.2015.10244
9. Rose AT, Patel RM. A critical analysis of risk factors for necrotizing enterocolitis. Semin Fetal Neonatal Med. (2018) 23:374–9. doi: 10.1016/j.siny.2018.07.005
10. Alsaied A, Islam N, Thalib L. Global incidence of Necrotizing Enterocolitis: a systematic review and meta-analysis. BMC Pediatr. (2020) 20:344. doi: 10.1186/s12887-020-02231-5
11. Claud EC, Lu L, Anton PM, Savidge T, Walker WA, Cherayil BJ. Developmentally regulated IkappaB expression in intestinal epithelium and susceptibility to flagellin-induced inflammation. Proc Natl Acad Sci USA. (2004) 101:7404–8. doi: 10.1073/pnas.0401710101
12. Walls Castellanos M, Claud EC. The microbiome, guard or threat to infant health. Trends Mol Med. (2021) 27:1175–86. doi: 10.1016/j.molmed.2021.08.002
13. Hunter CJ, Upperman JS, Ford HR, Camerini V. Understanding the susceptibility of the premature infant to necrotizing enterocolitis (NEC). Pediatr Res. (2008) 63:117–23. doi: 10.1203/PDR.0b013e31815ed64c
14. Li B, Wu RY, Horne RG, Ahmed A, Lee D, Robinson SC, et al. Human milk oligosaccharides protect against necrotizing enterocolitis by activating intestinal cell differentiation. Mol Nutr Food Res. (2020) 64:e2000519. doi: 10.1002/mnfr.202000519
15. Gopalakrishna KP, Macadangdang BR, Rogers MB, Tometich JT, Firek BA, Baker R, et al. Maternal IgA protects against the development of necrotizing enterocolitis in preterm infants. Nat Med. (2019) 25:1110–5. doi: 10.1038/s41591-019-0480-9
16. Pammi M, Suresh G. Enteral lactoferrin supplementation for prevention of sepsis and necrotizing enterocolitis in preterm infants. Cochrane Database Syst Rev. (2017) 6:CD007137. doi: 10.1002/14651858.CD007137.pub5
17. Weintraub AS, Ferrara L, Deluca L, Moshier E, Green RS, Oakman E, et al. Antenatal antibiotic exposure in preterm infants with necrotizing enterocolitis. J Perinatol. (2012) 32:705–9. doi: 10.1038/jp.2011.180
18. Wong D, Abdel-Latif M, Kent A, NICUS Network. Antenatal steroid exposure and outcomes of very premature infants: a regional cohort study. Arch Dis Child Fetal Neonatal Ed. (2014) 99:F12–F20. doi: 10.1136/archdischild-2013-304705
19. Lee J-Y, Park K-H, Kim A, Yang H-R, Jung E-Y, Cho S-H. Maternal and placental risk factors for developing necrotizing enterocolitis in very preterm infants. Pediatr Neonatol. (2017) 58:57–62. doi: 10.1016/j.pedneo.2016.01.005
20. Cetinkaya M, Ozkan H, Koksal N. Maternal preeclampsia is associated with increased risk of necrotizing enterocolitis in preterm infants. Early Hum Dev. (2012) 88:893–8. doi: 10.1016/j.earlhumdev.2012.07.004
21. Kamoji VM, Dorling JS, Manktelow B, Draper ES, Field DJ. Antenatal umbilical Doppler abnormalities: an independent risk factor for early onset neonatal necrotizing enterocolitis in premature infants. Acta Paediatr. (2008) 97:327–31. doi: 10.1111/j.1651-2227.2008.00671.x
22. Grandi C, Tapia JL, Cardoso VC. Impact of maternal diabetes mellitus on mortality and morbidity of very low birth weight infants: a multicenter Latin America study. J Pediatr (Rio J). (2015) 91:234–41. doi: 10.1016/j.jped.2014.08.007
23. Guthrie SO, Gordon PV, Thomas V, Thorp JA, Peabody J, Clark RH. Necrotizing enterocolitis among neonates in the United States. J Perinatol. (2003) 23:278–85. doi: 10.1038/sj.jp.7210892
24. Son M, Grobman WA, Miller ES. Is mode of delivery associated with the risk of necrotizing enterocolitis? Am J Obstet Gynecol. (2016) 215:389.e1–4. doi: 10.1016/j.ajog.2016.04.058
25. Battersby C, Longford N, Costeloe K, Modi N, UK Neonatal Collaborative Necrotising Enterocolitis Study Group. Development of a gestational age-specific case definition for neonatal necrotizing enterocolitis. JAMA Pediatr. (2017) 171:256–63. doi: 10.1001/jamapediatrics.2016.3633
26. Bardin C, Zelkowitz P, Papageorgiou A. Outcome of small-for-gestational age and appropriate-for-gestational age infants born before 27 weeks of gestation. Pediatrics. (1997) 100:E4. doi: 10.1542/peds.100.2.e4
27. Drenckpohl D, Knaub L, Schneider C, McConnell C, Huaping W, Macwan K. Risk factors that may predispose premature infants to increased incidence of necrotizing enterocolitis. Infant Child Adolesc Nutr. (2010) 2:37–44. doi: 10.1177/1941406409359195
28. Ree IMC, Smits-Wintjens VEHJ, Rijntjes-Jacobs EGJ, Pelsma ICM, Steggerda SJ, Walther FJ, et al. Necrotizing enterocolitis in small-for-gestational-age neonates: a matched case-control study. Neonatology. (2014) 105:74–8. doi: 10.1159/000356033
29. Frazer LC, Good M. Intestinal epithelium in early life. Mucosal Immunol. (2022) 15(6):1181–7. doi: 10.1038/s41385-022-00579-8
30. Shulhan J, Dicken B, Hartling L, Larsen BM. Current knowledge of necrotizing enterocolitis in preterm infants and the impact of different types of enteral nutrition products. Adv Nutr. (2017) 8:80–91. doi: 10.3945/an.116.013193
31. Siggers RH, Siggers J, Thymann T, Boye M, Sangild PT. Nutritional modulation of the gut microbiota and immune system in preterm neonates susceptible to necrotizing enterocolitis. J Nutr Biochem. (2011) 22:511–21. doi: 10.1016/j.jnutbio.2010.08.002
32. Lucas A, Cole TJ. Breast milk and neonatal necrotising enterocolitis. Lancet. (1990) 336:1519–23. doi: 10.1016/0140-6736(90)93304-8
33. Altobelli E, Angeletti PM, Verrotti A, Petrocelli R. The impact of human milk on necrotizing enterocolitis: a systematic review and meta-analysis. Nutrients. (2020) 12(5):1322. doi: 10.3390/nu12051322
34. Schanler RJ, Shulman RJ, Lau C. Feeding strategies for premature infants: beneficial outcomes of feeding fortified human milk versus preterm formula. Pediatrics. (1999) 103:1150–7. doi: 10.1542/peds.103.6.1150
35. Carlisle EM, Morowitz MJ. The intestinal microbiome and necrotizing enterocolitis. Curr Opin Pediatr. (2013) 25:382–7. doi: 10.1097/MOP.0b013e3283600e91
36. Esmaeilizand R, Shah PS, Seshia M, Yee W, Yoon EW, Dow K, et al. Antibiotic exposure and development of necrotizing enterocolitis in very preterm neonates. Paediatr Child Health. (2018) 23:e56–e61. doi: 10.1093/pch/pxx169
37. Lu P, Sodhi CP, Hackam DJ. Toll-like receptor regulation of intestinal development and inflammation in the pathogenesis of necrotizing enterocolitis. Pathophysiology. (2014) 21:81–93. doi: 10.1016/j.pathophys.2013.11.007
38. Cho SX, Berger PJ, Nold-Petry CA, Nold MF. The immunological landscape in necrotising enterocolitis. Expert Rev Mol Med. (2016) 18:e12. doi: 10.1017/erm.2016.13
39. Olm MR, Bhattacharya N, Crits-Christoph A, Firek BA, Baker R, Song YS, et al. Necrotizing enterocolitis is preceded by increased gut bacterial replication, Klebsiella, and fimbriae-encoding bacteria. Sci Adv. (2019) 5:eaax5727. doi: 10.1126/sciadv.aax5727
40. Healy DB, Ryan CA, Ross RP, Stanton C, Dempsey EM. Clinical implications of preterm infant gut microbiome development. Nat Microbiol. (2022) 7:22–33. doi: 10.1038/s41564-021-01025-4
41. Hackam DJ, Sodhi CP. Bench to bedside—new insights into the pathogenesis of necrotizing enterocolitis. Nat Rev Gastroenterol Hepatol. (2022) 19:468–79. doi: 10.1038/s41575-022-00594-x
42. Henneke P, Kierdorf K, Hall LJ, Sperandio M, Hornef M. Perinatal development of innate immune topology. eLife. (2021) 10:e67793. doi: 10.7554/eLife.67793
43. Malek A, Sager R, Kuhn P, Nicolaides KH, Schneider H. Evolution of maternofetal transport of immunoglobulins during human pregnancy. Am J Reprod Immunol. (1996) 36:248–55. doi: 10.1111/j.1600-0897.1996.tb00172.x
44. Sanidad KZ, Amir M, Ananthanarayanan A, Singaraju A, Shiland NB, Hong HS, et al. Maternal gut microbiome-induced IgG regulates neonatal gut microbiome and immunity. Sci Immunol. (2022) 7:eabh3816. doi: 10.1126/sciimmunol.abh3816
45. Boudry G, Charton E, Le Huerou-Luron I, Ferret-Bernard S, Le Gall S, Even S, et al. The relationship between breast milk components and the infant gut microbiota. Front Nutr. (2021) 8:629740. doi: 10.3389/fnut.2021.629740
46. Abbas S, Keir AK, Makrides M, Klein LD, Grzeskowiak LE, McPhee AJ, et al. Tailoring human milk oligosaccharides to prevent necrotising enterocolitis among preterm infants. Front Nutr. (2021) 8:702888. doi: 10.3389/fnut.2021.702888
47. Ninonuevo MR, Park Y, Yin H, Zhang J, Ward RE, Clowers BH, et al. A strategy for annotating the human milk glycome. J Agric Food Chem. (2006) 54:7471–80. doi: 10.1021/jf0615810
48. Plaza-Díaz J, Fontana L, Gil A. Human milk oligosaccharides and immune system development. Nutrients. (2018) 10(8):1038. doi: 10.3390/nu10081038
49. Marcobal A, Barboza M, Froehlich JW, Block DE, German JB, Lebrilla CB, et al. Consumption of human milk oligosaccharides by gut-related microbes. J Agric Food Chem. (2010) 58:5334–40. doi: 10.1021/jf9044205
50. Marcobal A, Sonnenburg JL. Human milk oligosaccharide consumption by intestinal microbiota. Clin Microbiol Infect. (2012) 18(Suppl 4):12–5. doi: 10.1111/j.1469-0691.2012.03863.x
51. Sodhi CP, Wipf P, Yamaguchi Y, Fulton WB, Kovler M, Niño DF, et al. The human milk oligosaccharides 2′-fucosyllactose and 6′-sialyllactose protect against the development of necrotizing enterocolitis by inhibiting toll-like receptor 4 signaling. Pediatr Res. (2021) 89:91–101. doi: 10.1038/s41390-020-0852-3
52. Wang Y, Zou Y, Wang J, Ma H, Zhang B, Wang S. The protective effects of 2′-fucosyllactose against E. Coli O157 infection are mediated by the regulation of gut microbiota and the inhibition of pathogen adhesion. Nutrients. (2020) 12(5):1284. doi: 10.3390/nu12051284
53. Masi AC, Embleton ND, Lamb CA, Young G, Granger CL, Najera J, et al. Human milk oligosaccharide DSLNT and gut microbiome in preterm infants predicts necrotising enterocolitis. Gut. (2021) 70:2273–82. doi: 10.1136/gutjnl-2020-322771
54. Wu RY, Li B, Koike Y, Määttänen P, Miyake H, Cadete M, et al. Human milk oligosaccharides increase mucin expression in experimental necrotizing enterocolitis. Mol Nutr Food Res. (2019) 63:e1800658. doi: 10.1002/mnfr.201800658
55. Jakaitis BM, Denning PW. Human breast milk and the gastrointestinal innate immune system. Clin Perinatol. (2014) 41:423–35. doi: 10.1016/j.clp.2014.02.011
56. Plaisancié P, Boutrou R, Estienne M, Henry G, Jardin J, Paquet A, et al. β-Casein(94-123)-derived peptides differently modulate production of mucins in intestinal goblet cells. J Dairy Res. (2015) 82:36–46. doi: 10.1017/S0022029914000533
57. Moya FR, Eguchi H, Zhao B, Furukawa M, Sfeir J, Osorio M, et al. Platelet-activating factor acetylhydrolase in term and preterm human milk: a preliminary report. J Pediatr Gastroenterol Nutr. (1994) 19:236–9. doi: 10.1097/00005176-199408000-00015
58. Emami CN, Chokshi N, Wang J, Hunter C, Guner Y, Goth K, et al. Role of interleukin-10 in the pathogenesis of necrotizing enterocolitis. Am J Surg. (2012) 203:428–35. doi: 10.1016/j.amjsurg.2011.08.016
59. Frost BL, Jilling T, Lapin B, Maheshwari A, Caplan MS. Maternal breast milk transforming growth factor-beta and feeding intolerance in preterm infants. Pediatr Res. (2014) 76:386–93. doi: 10.1038/pr.2014.96
60. Good M, Sodhi CP, Egan CE, Afrazi A, Jia H, Yamaguchi Y, et al. Breast milk protects against the development of necrotizing enterocolitis through inhibition of Toll-like receptor 4 in the intestinal epithelium via activation of the epidermal growth factor receptor. Mucosal Immunol. (2015) 8:1166–79. doi: 10.1038/mi.2015.30
61. Knoop KA, Coughlin PE, Floyd AN, Ndao IM, Hall-Moore C, Shaikh N, et al. Maternal activation of the EGFR prevents translocation of gut-residing pathogenic Escherichia coli in a model of late-onset neonatal sepsis. Proc Natl Acad Sci USA. (2020) 117:7941–9. doi: 10.1073/pnas.1912022117
62. Good M, Siggers RH, Sodhi CP, Afrazi A, Alkhudari F, Egan CE, et al. Amniotic fluid inhibits Toll-like receptor 4 signaling in the fetal and neonatal intestinal epithelium. Proc Natl Acad Sci USA. (2012) 109:11330–5. doi: 10.1073/pnas.1200856109
63. Yan X, Managlia E, Zhao Y-Y, Tan X-D, De Plaen IG. Macrophage-derived IGF-1 protects the neonatal intestine against necrotizing enterocolitis by promoting microvascular development. Commun Biol. (2022) 5:320. doi: 10.1038/s42003-022-03252-9
64. Holgersen K, Rasmussen MB, Carey G, Burrin DG, Thymann T, Sangild PT. Clinical outcome and gut development after insulin-like growth factor-1 supplementation to preterm pigs. Front Pediatr. (2022) 10:868911. doi: 10.3389/fped.2022.868911
65. Holgersen K, Gao X, Narayanan R, Gaur T, Carey G, Barton N, et al. Supplemental insulin-like growth factor-1 and necrotizing enterocolitis in preterm pigs. Front Pediatr. (2020) 8:602047. doi: 10.3389/fped.2020.602047
66. Johansson MEV, Hansson GC. Immunological aspects of intestinal mucus and mucins. Nat Rev Immunol. (2016) 16:639–49. doi: 10.1038/nri.2016.88
67. Vieten D, Corfield A, Carroll D, Ramani P, Spicer R. Impaired mucosal regeneration in neonatal necrotising enterocolitis. Pediatr Surg Int. (2005) 21:153–60. doi: 10.1007/s00383-004-1312-6
68. Hall T, Dymock D, Corfield AP, Weaver G, Woodward M, Berry M. Bacterial invasion of HT29-MTX-E12 monolayers: effects of human breast milk. J Pediatr Surg. (2013) 48:353–7; discussion 357. doi: 10.1016/j.jpedsurg.2012.11.021
69. Engevik MA, Yacyshyn MB, Engevik KA, Wang J, Darien B, Hassett DJ, et al. Human Clostridium difficile infection: altered mucus production and composition. Am J Physiol Gastrointest Liver Physiol. (2015) 308:G510–24. doi: 10.1152/ajpgi.00091.2014
70. Le C-F, Fang C-M, Sekaran SD. Intracellular targeting mechanisms by antimicrobial peptides. Antimicrob Agents Chemother. (2017) 61(4):e02340-16. doi: 10.1128/AAC.02340-16
71. Ma Y, Guo Z, Xia B, Zhang Y, Liu X, Yu Y, et al. Identification of antimicrobial peptides from the human gut microbiome using deep learning. Nat Biotechnol. (2022) 40:921–31. doi: 10.1038/s41587-022-01226-0
72. Liang W, Enée E, Andre-Vallee C, Falcone M, Sun J, Diana J. Intestinal cathelicidin antimicrobial peptide shapes a protective neonatal gut microbiota against pancreatic autoimmunity. Gastroenterology. (2022) 162:1288–302.e16. doi: 10.1053/j.gastro.2021.12.272
73. Underwood MA, Kananurak A, Coursodon CF, Adkins-Reick CK, Chu H, Bennett SH, et al. Bifidobacterium bifidum in a rat model of necrotizing enterocolitis: antimicrobial peptide and protein responses. Pediatr Res. (2012) 71:546–51. doi: 10.1038/pr.2012.11
74. Sheng Q, Lv Z, Cai W, Song H, Qian L, Mu H, et al. Human β-defensin-3 promotes intestinal epithelial cell migration and reduces the development of necrotizing enterocolitis in a neonatal rat model. Pediatr Res. (2014) 76:269–79. doi: 10.1038/pr.2014.93
75. Killick J, Morisse G, Sieger D, Astier AL. Complement as a regulator of adaptive immunity. Semin Immunopathol. (2018) 40:37–48. doi: 10.1007/s00281-017-0644-y
76. Singh DK, Tóth R, Gácser A. Mechanisms of pathogenic candida species to evade the host complement attack. Front Cell Infect Microbiol. (2020) 10:94. doi: 10.3389/fcimb.2020.00094
77. Notarangelo LD, Chirico G, Chiara A, Colombo A, Rondini G, Plebani A, et al. Activity of classical and alternative pathways of complement in preterm and small for gestational age infants. Pediatr Res. (1984) 18:281–5. doi: 10.1203/00006450-198403000-00014
78. Grumach AS, Ceccon ME, Rutz R, Fertig A, Kirschfink M. Complement profile in neonates of different gestational ages. Scand J Immunol. (2014) 79:276–81. doi: 10.1111/sji.12154
79. Johnston RB, Altenburger KM, Atkinson AW, Curry RH. Complement in the newborn infant. Pediatrics. (1979) 64:781–6. doi: 10.1542/peds.64.5.781
80. Høgåsen AK, Overlie I, Hansen TW, Abrahamsen TG, Finne PH, Høgåsen K. The analysis of the complement activation product SC5 b-9 is applicable in neonates in spite of their profound C9 deficiency. J Perinat Med. (2000) 28:39–48. doi: 10.1515/JPM.2000.006
81. Tayman C, Tonbul A, Kahveci H, Uysal S, Koseoğlu B, Tatli MM, et al. C5a, a complement activation product, is a useful marker in predicting the severity of necrotizing enterocolitis. Tohoku J Exp Med. (2011) 224:143–50. doi: 10.1620/tjem.224.143
82. Schlapbach LJ, Aebi C, Fisch U, Ammann RA, Otth M, Bigler S, et al. Higher cord blood levels of mannose-binding lectin-associated serine protease-2 in infants with necrotising enterocolitis. Pediatr Res. (2008) 64:562–6. doi: 10.1203/PDR.0b013e3181841335
83. Sampah MES, Hackam DJ. Dysregulated mucosal immunity and associated pathogeneses in preterm neonates. Front Immunol. (2020) 11:899. doi: 10.3389/fimmu.2020.00899
84. Zhao X, Liang W, Wang Y, Yi R, Luo L, Wang W, et al. Ontogeny of RORγt+ cells in the intestine of newborns and its role in the development of experimental necrotizing enterocolitis. Cell Biosci. (2022) 12:3. doi: 10.1186/s13578-021-00739-6
85. Weitkamp J-H, Rudzinski E, Koyama T, Correa H, Matta P, Alberty B, et al. Ontogeny of FOXP3(+) regulatory T cells in the postnatal human small intestinal and large intestinal lamina propria. Pediatr Dev Pathol. (2009) 12:443–9. doi: 10.2350/08-09-0533.1
86. Zuiderwijk MO, van der Burg M, Bekker V, Schoenaker MHD. Regulatory T cells in development and prediction of necrotizing enterocolitis in preterm neonates: a scoping review. Int J Mol Sci. (2022) 23(18):10903. doi: 10.3390/ijms231810903
87. Hourigan SK, Ta A, Wong WSW, Clemency NC, Provenzano MG, Baveja R, et al. The microbiome in necrotizing enterocolitis: a case report in twins and minireview. Clin Ther. (2016) 38:747–53. doi: 10.1016/j.clinthera.2016.02.014
88. Denning N-L, Prince JM. Neonatal intestinal dysbiosis in necrotizing enterocolitis. Mol Med. (2018) 24:4. doi: 10.1186/s10020-018-0002-0
89. Dingle BM, Liu Y, Fatheree NY, Min J, Rhoads JM, Tran DQ. Foxp3+ regulatory T cells attenuate experimental necrotizing enterocolitis. PLoS ONE. (2013) 8:e82963. doi: 10.1371/journal.pone.0082963
90. Aziz M, Prince JM, Wang P. Gut microbiome and necrotizing enterocolitis: understanding the connection to find a cure. Cell Host Microbe. (2022) 30:612–6. doi: 10.1016/j.chom.2022.04.003
91. Wang Q, Dong J, Zhu Y. Probiotic supplement reduces risk of necrotizing enterocolitis and mortality in preterm very low-birth-weight infants: an updated meta-analysis of 20 randomized, controlled trials. J Pediatr Surg. (2012) 47:241–8. doi: 10.1016/j.jpedsurg.2011.09.064
92. Chang H-Y, Chen J-H, Chang J-H, Lin H-C, Lin C-Y, Peng C-C. Multiple strains probiotics appear to be the most effective probiotics in the prevention of necrotizing enterocolitis and mortality: an updated meta-analysis. PLoS ONE. (2017) 12:e0171579. doi: 10.1371/journal.pone.0171579
93. Robertson C, Savva GM, Clapuci R, Jones J, Maimouni H, Brown E, et al. Incidence of necrotising enterocolitis before and after introducing routine prophylactic Lactobacillus and Bifidobacterium probiotics. Arch Dis Child Fetal Neonatal Ed. (2020) 105:380–6. doi: 10.1136/archdischild-2019-317346
94. Wang Y, Hoenig JD, Malin KJ, Qamar S, Petrof EO, Sun J, et al. 16S rRNA gene-based analysis of fecal microbiota from preterm infants with and without necrotizing enterocolitis. ISME J. (2009) 3:944–54. doi: 10.1038/ismej.2009.37
95. Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci USA. (2010) 107:11971–5. doi: 10.1073/pnas.1002601107
96. Smith PD, Smythies LE, Shen R, Greenwell-Wild T, Gliozzi M, Wahl SM. Intestinal macrophages and response to microbial encroachment. Mucosal Immunol. (2011) 4:31–42. doi: 10.1038/mi.2010.66
97. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. (2017) 17:349–62. doi: 10.1038/nri.2017.28
98. Pang Y, Du X, Xu X, Wang M, Li Z. Monocyte activation and inflammation can exacerbate Treg/Th17 imbalance in infants with neonatal necrotizing enterocolitis. Int Immunopharmacol. (2018) 59:354–60. doi: 10.1016/j.intimp.2018.04.026
99. Managlia E, Liu SXL, Yan X, Tan X-D, Chou PM, Barrett TA, et al. Blocking NF-κB activation in Ly6c+ monocytes attenuates necrotizing enterocolitis. Am J Pathol. (2019) 189:604–18. doi: 10.1016/j.ajpath.2018.11.015
100. Olaloye OO, Liu P, Toothaker JM, McCourt BT, McCourt CC, Xiao J, et al. CD16+CD163+ Monocytes traffic to sites of inflammation during necrotizing enterocolitis in premature infants. J Exp Med. (2021) 218(9):e20200344. doi: 10.1084/jem.20200344
101. Remon J, Kampanatkosol R, Kaul RR, Muraskas JK, Christensen RD, Maheshwari A. Acute drop in blood monocyte count differentiates NEC from other causes of feeding intolerance. J Perinatol. (2014) 34:549–54. doi: 10.1038/jp.2014.52
102. Wei J, Besner GE. M1 to M2 macrophage polarization in heparin-binding epidermal growth factor-like growth factor therapy for necrotizing enterocolitis. J Surg Res. (2015) 197:126–38. doi: 10.1016/j.jss.2015.03.023
103. Wei J, Tang D, Lu C, Yang J, Lu Y, Wang Y, et al. Irf5 deficiency in myeloid cells prevents necrotizing enterocolitis by inhibiting M1 macrophage polarization. Mucosal Immunol. (2019) 12:888–96. doi: 10.1038/s41385-019-0169-x
104. Nauseef WM, Borregaard N. Neutrophils at work. Nat Immunol. (2014) 15:602–11. doi: 10.1038/ni.2921
105. Zhang T, Jiang J, Liu J, Xu L, Duan S, Sun L, et al. MK2 Is required for neutrophil-derived ROS production and inflammatory bowel disease. Front Med (Lausanne). (2020) 7:207. doi: 10.3389/fmed.2020.00207
106. Christensen RD, Yoder BA, Baer VL, Snow GL, Butler A. Early-onset neutropenia in small-for-gestational-age infants. Pediatrics. (2015) 136:e1259–67. doi: 10.1542/peds.2015-1638
107. Zindl CL, Lai J-F, Lee YK, Maynard CL, Harbour SN, Ouyang W, et al. IL-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc Natl Acad Sci USA. (2013) 110:12768–73. doi: 10.1073/pnas.1300318110
108. Mihi B, Gong Q, Nolan LS, Gale SE, Goree M, Hu E, et al. Interleukin-22 signaling attenuates necrotizing enterocolitis by promoting epithelial cell regeneration. Cell Rep Med. (2021) 2:100320. doi: 10.1016/j.xcrm.2021.100320
109. Emami CN, Mittal R, Wang L, Ford HR, Prasadarao NV. Role of neutrophils and macrophages in the pathogenesis of necrotizing enterocolitis caused by Cronobacter sakazakii. J Surg Res. (2012) 172:18–28. doi: 10.1016/j.jss.2011.04.019
110. Chen Z, Zhang Y, Lin R, Meng X, Zhao W, Shen W, et al. Cronobacter sakazakii induces necrotizing enterocolitis by regulating NLRP3 inflammasome expression via TLR4. J Med Microbiol. (2020) 69:748–58. doi: 10.1099/jmm.0.001181
111. Nolan LS, Mihi B, Agrawal P, Gong Q, Rimer JM, Bidani SS, et al. Indole-3-carbinol-dependent aryl hydrocarbon receptor signaling attenuates the inflammatory response in experimental necrotizing enterocolitis. Immunohorizons. (2021) 5:193–209. doi: 10.4049/immunohorizons.2100018
112. Helander HF, Fändriks L. Surface area of the digestive tract—revisited. Scand J Gastroenterol. (2014) 49:681–9. doi: 10.3109/00365521.2014.898326
113. Poussier P, Edouard P, Lee C, Binnie M, Julius M. Thymus-independent development and negative selection of T cells expressing T cell receptor alpha/beta in the intestinal epithelium: evidence for distinct circulation patterns of gut- and thymus-derived T lymphocytes. J Exp Med. (1992) 176:187–99. doi: 10.1084/jem.176.1.187
114. McDonald BD, Jabri B, Bendelac A. Diverse developmental pathways of intestinal intraepithelial lymphocytes. Nat Rev Immunol. (2018) 18:514–25. doi: 10.1038/s41577-018-0013-7
115. Inagaki-Ohara K, Chinen T, Matsuzaki G, Sasaki A, Sakamoto Y, Hiromatsu K, et al. Mucosal T cells bearing TCRgammadelta play a protective role in intestinal inflammation. J Immunol. (2004) 173:1390–8. doi: 10.4049/jimmunol.173.2.1390
116. Weitkamp J-H, Rosen MJ, Zhao Z, Koyama T, Geem D, Denning TL, et al. Small intestinal intraepithelial TCRγδ+ T lymphocytes are present in the premature intestine but selectively reduced in surgical necrotizing enterocolitis. PLoS ONE. (2014) 9:e99042. doi: 10.1371/journal.pone.0099042
117. Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, et al. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell. (2011) 147:629–40. doi: 10.1016/j.cell.2011.09.025
118. Lu P, Yamaguchi Y, Fulton WB, Wang S, Zhou Q, Jia H, et al. Maternal aryl hydrocarbon receptor activation protects newborns against necrotizing enterocolitis. Nat Commun. (2021) 12:1042. doi: 10.1038/s41467-021-21356-4
119. Rhoads JM. Protective function of FoxP3 regulatory T cells in experimental necrotizing enterocolitis (P1010). J Immunol. (2013) 190(1_Supplement):65.6. doi: 10.4049/jimmunol.190.Supp.65.6
120. Weitkamp J-H, Koyama T, Rock MT, Correa H, Goettel JA, Matta P, et al. Necrotising enterocolitis is characterised by disrupted immune regulation and diminished mucosal regulatory (FOXP3)/effector (CD4, CD8) T cell ratios. Gut. (2013) 62:73–82. doi: 10.1136/gutjnl-2011-301551
121. Pang Y, Du X, Xu X, Wang M, Li Z. Impairment of regulatory T cells in patients with neonatal necrotizing enterocolitis. Int Immunopharmacol. (2018) 63:19–25. doi: 10.1016/j.intimp.2018.07.029
122. Egan CE, Sodhi CP, Good M, Lin J, Jia H, Yamaguchi Y, et al. Toll-like receptor 4-mediated lymphocyte influx induces neonatal necrotizing enterocolitis. J Clin Invest. (2016) 126:495–508. doi: 10.1172/JCI83356
123. Niño DF, Sodhi CP, Egan CE, Zhou Q, Lin J, Lu P, et al. Retinoic acid improves incidence and severity of necrotizing enterocolitis by lymphocyte balance restitution and repopulation of LGR5+ intestinal stem cells. Shock. (2017) 47:22–32. doi: 10.1097/SHK.0000000000000713
124. Servin AL. Antagonistic activities of lactobacilli and bifidobacteria against microbial pathogens. FEMS Microbiol Rev. (2004) 28:405–40. doi: 10.1016/j.femsre.2004.01.003
125. Spaggiari L, Sala A, Ardizzoni A, De Seta F, Singh DK, Gacser A, et al. Lactobacillus acidophilus, L. plantarum, L. rhamnosus, and L. reuteri cell-free supernatants inhibit Candida parapsilosis pathogenic potential upon infection of vaginal epithelial cells monolayer and in a transwell coculture system in vitro. Microbiol Spectr. (2022) 10:e0269621. doi: 10.1128/spectrum.02696-21
126. Long G, Hu Y, Tao E, Chen B, Shu X, Zheng W, et al. The influence of cesarean section on the composition and development of gut microbiota during the first 3 months of life. Front Microbiol. (2021) 12:691312. doi: 10.3389/fmicb.2021.691312
127. Bäckhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. (2015) 17:690–703. doi: 10.1016/j.chom.2015.04.004
128. Schönherr-Hellec S, Klein GL, Delannoy J, Ferraris L, Rozé JC, Butel MJ, et al. Clostridial strain-specific characteristics associated with necrotizing enterocolitis. Appl Environ Microbiol. (2018) 84(7):e02428-17. doi: 10.1128/AEM.02428-17
129. Schönherr-Hellec S, Aires J. Clostridia and necrotizing enterocolitis in preterm neonates. Anaerobe. (2019) 58:6–12. doi: 10.1016/j.anaerobe.2019.04.005
130. Geil CC, Castle WK, Mortimer EA. Group A streptococcal infections in newborn nurseries. Pediatrics. (1970) 46:849–54. doi: 10.1542/peds.46.6.849
131. Coudron PE, Mayhall CG, Facklam RR, Spadora AC, Lamb VA, Lybrand MR, et al. Streptococcus faecium outbreak in a neonatal intensive care unit. J Clin Microbiol. (1984) 20:1044–8. doi: 10.1128/jcm.20.6.1044-1048.1984
132. Luginbuhl LM, Rotbart HA, Facklam RR, Roe MH, Elliot JA. Neonatal enterococcal sepsis: case-control study and description of an outbreak. Pediatr Infect Dis J. (1987) 6:1022–6. doi: 10.1097/00006454-198706110-00003
133. Heath JA, Zerr DM. Infections acquired in the nursery: epidemiology and control. In: Infectious diseases of the fetus and newborn infant. Elsevier. (2006). p. 1179–205. doi: 10.1016/B0-72-160537-0/50037-2
134. Scheifele DW, Bjornson GL, Dyer RA, Dimmick JE. Delta-like toxin produced by coagulase-negative staphylococci is associated with neonatal necrotizing enterocolitis. Infect Immun. (1987) 55:2268–73. doi: 10.1128/iai.55.9.2268-2273.1987
135. Sim K, Shaw AG, Randell P, Cox MJ, McClure ZE, Li M-S, et al. Dysbiosis anticipating necrotizing enterocolitis in very premature infants. Clin Infect Dis. (2015) 60:389–97. doi: 10.1093/cid/ciu822
136. Cheng C, He Y, Xiao S, Ai Q, Yu J. The association between enteric viruses and necrotizing enterocolitis. Eur J Pediatr. (2021) 180:225–32. doi: 10.1007/s00431-020-03746-w
137. Kaelin EA, Rodriguez C, Hall-Moore C, Hoffmann JA, Linneman LA, Ndao IM, et al. Longitudinal gut virome analysis identifies specific viral signatures that precede necrotizing enterocolitis onset in preterm infants. Nat Microbiol. (2022) 7:653–62. doi: 10.1038/s41564-022-01096-x
138. Ralls MW, Gadepalli SK, Sylvester KG, Good M. Development of the necrotizing enterocolitis society registry and biorepository. Semin Pediatr Surg. (2018) 27:25–8. doi: 10.1053/j.sempedsurg.2017.11.005
139. Mendez YS, Khan FA, Perrier GV, Radulescu A. Animal models of necrotizing enterocolitis. World Jnl Ped Surgery. (2020) 3:e000109. doi: 10.1136/wjps-2020-000109
140. Caplan MS, Hedlund E, Adler L, Hsueh W. Role of asphyxia and feeding in a neonatal rat model of necrotizing enterocolitis. Pediatr Pathol. (1994) 14:1017–28. doi: 10.3109/15513819409037698
141. Barlow B, Santulli TV. Importance of multiple episodes of hypoxia or cold stress on the development of enterocolitis in an animal model. Surgery. (1975) 77:687–90. PMID: 1173200
142. Ares GJ, McElroy SJ, Hunter CJ. The science and necessity of using animal models in the study of necrotizing enterocolitis. Semin Pediatr Surg. (2018) 27:29–33. doi: 10.1053/j.sempedsurg.2017.11.006
143. Lu P, Sodhi CP, Jia H, Shaffiey S, Good M, Branca MF, et al. Animal models of gastrointestinal and liver diseases. Animal models of necrotizing enterocolitis: pathophysiology, translational relevance, and challenges. Am J Physiol Gastrointest Liver Physiol. (2014) 306:G917–28. doi: 10.1152/ajpgi.00422.2013
144. Charreau B, Tesson L, Soulillou JP, Pourcel C, Anegon I. Transgenesis in rats: technical aspects and models. Transgenic Res. (1996) 5:223–34. doi: 10.1007/BF01972876
145. Nolan LS, Gong Q, Hofmeister HN, Good M. A protocol for the induction of experimental necrotizing enterocolitis in neonatal mice. STAR Protocols. (2021) 2:100951. doi: 10.1016/j.xpro.2021.100951
146. Niño DF, Zhou Q, Yamaguchi Y, Martin LY, Wang S, Fulton WB, et al. Cognitive impairments induced by necrotizing enterocolitis can be prevented by inhibiting microglial activation in mouse brain. Sci Transl Med. (2018) 10(471):eaan0237. doi: 10.1126/scitranslmed.aan0237
147. McCarthy R, Martin-Fairey C, Sojka DK, Herzog ED, Jungheim ES, Stout MJ, et al. Mouse models of preterm birth: suggested assessment and reporting guidelines. Biol Reprod. (2018) 99:922–37. doi: 10.1093/biolre/ioy109
148. Call L, Stoll B, Oosterloo B, Ajami N, Sheikh F, Wittke A, et al. Metabolomic signatures distinguish the impact of formula carbohydrates on disease outcome in a preterm piglet model of NEC. Microbiome. (2018) 6:111. doi: 10.1186/s40168-018-0498-0
149. Hui Y, Vestergaard G, Deng L, Kot WP, Thymann T, Brunse A, et al. Donor-dependent fecal microbiota transplantation efficacy against necrotizing enterocolitis in preterm pigs. NPJ Biofilms Microbiomes. (2022) 8:48. doi: 10.1038/s41522-022-00310-2
150. Sangild PT, Siggers RH, Schmidt M, Elnif J, Bjornvad CR, Thymann T, et al. Diet- and colonization-dependent intestinal dysfunction predisposes to necrotizing enterocolitis in preterm pigs. Gastroenterology. (2006) 130:1776–92. doi: 10.1053/j.gastro.2006.02.026
151. Sulistyo A, Rahman A, Biouss G, Antounians L, Zani A. Animal models of necrotizing enterocolitis: review of the literature and state of the art. Innov Surg Sci. (2018) 3:87–92. doi: 10.1515/iss-2017-0050
152. Siggers RH, Thymann T, Jensen BB, Mølbak L, Heegaard PMH, Schmidt M, et al. Elective cesarean delivery affects gut maturation and delays microbial colonization but does not increase necrotizing enterocolitis in preterm pigs. Am J Physiol Regul Integr Comp Physiol. (2008) 294:R929–38. doi: 10.1152/ajpregu.00705.2007
153. De Fazio L, Beghetti I, Bertuccio SN, Marsico C, Martini S, Masetti R, et al. Necrotizing enterocolitis: overview on in vitro models. Int J Mol Sci. (2021) 22(13):6761. doi: 10.3390/ijms22136761
154. Pimenta J, Ribeiro R, Almeida R, Costa PF, da Silva MA, Pereira B. Organ-on-chip approaches for intestinal 3D in vitro modeling. Cell Mol Gastroenterol Hepatol. (2022) 13:351–67. doi: 10.1016/j.jcmgh.2021.08.015
155. Donkers JM, Eslami Amirabadi H, van de Steeg E. Intestine-on-a-chip: next level in vitro research model of the human intestine. Curr Opin Toxicol. (2021) 25:6–14. doi: 10.1016/j.cotox.2020.11.002
156. Bozzetti V, Senger S. Organoid technologies for the study of intestinal microbiota-host interactions. Trends Mol Med. (2022) 28:290–303. doi: 10.1016/j.molmed.2022.02.001
157. Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. (2009) 459:262–5. doi: 10.1038/nature07935
158. Ashammakhi N, Nasiri R, de Barros NR, Tebon P, Thakor J, Goudie M, et al. Gut-on-a-chip: current progress and future opportunities. Biomaterials. (2020) 255:120196. doi: 10.1016/j.biomaterials.2020.120196
159. Xiang Y, Wen H, Yu Y, Li M, Fu X, Huang S. Gut-on-chip: recreating human intestine in vitro. J Tissue Eng. (2020) 11:2041731420965318. doi: 10.1177/2041731420965318
160. Poletti M, Arnauts K, Ferrante M, Korcsmaros T. Organoid-based models to study the role of host-microbiota interactions in IBD. J Crohns Colitis. (2021) 15:1222–35. doi: 10.1093/ecco-jcc/jjaa257
161. Noel G, Baetz NW, Staab JF, Donowitz M, Kovbasnjuk O, Pasetti MF, et al. A primary human macrophage-enteroid co-culture model to investigate mucosal gut physiology and host-pathogen interactions. Sci Rep. (2017) 7:45270. doi: 10.1038/srep45270
162. Sasaki N, Miyamoto K, Maslowski KM, Ohno H, Kanai T, Sato T. Development of a scalable coculture system for gut anaerobes and human colon epithelium. Gastroenterology. (2020) 159:388–90.e5. doi: 10.1053/j.gastro.2020.03.021
163. Sato T, Stange DE, Ferrante M, Vries RGJ, Van Es JH, Van den Brink S, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. (2011) 141:1762–72. doi: 10.1053/j.gastro.2011.07.050
164. Co JY, Margalef-Català M, Li X, Mah AT, Kuo CJ, Monack DM, et al. Controlling epithelial polarity: a human enteroid model for host-pathogen interactions. Cell Rep. (2019) 26:2509–20.e4. doi: 10.1016/j.celrep.2019.01.108
165. Wilson SS, Mayo M, Melim T, Knight H, Patnaude L, Wu X, et al. Optimized culture conditions for improved growth and functional differentiation of mouse and human colon organoids. Front Immunol. (2020) 11:547102. doi: 10.3389/fimmu.2020.547102
166. Kakni P, López-Iglesias C, Truckenmüller R, Habibović P, Giselbrecht S. Reversing epithelial polarity in pluripotent stem cell-derived intestinal organoids. Front Bioeng Biotechnol. (2022) 10:879024. doi: 10.3389/fbioe.2022.879024
167. Mateer SW, Cardona J, Marks E, Goggin BJ, Hua S, Keely S. Ex vivo intestinal sacs to assess mucosal permeability in models of gastrointestinal disease. J Vis Exp. (2016) (108):e53250. doi: 10.3791/53250
168. Ladd MR, Costello CM, Gosztyla C, Werts AD, Johnson B, Fulton WB, et al. Development of intestinal scaffolds that mimic native mammalian intestinal tissue. Tissue Eng Part A. (2019) 25:1225–41. doi: 10.1089/ten.TEA.2018.0239
169. de Hoyos-Vega JM, Gonzalez-Suarez AM, Garcia-Cordero JL. A versatile microfluidic device for multiple ex vivo/in vitro tissue assays unrestrained from tissue topography. Microsyst Nanoeng. (2020) 6:40. doi: 10.1038/s41378-020-0156-0
170. Stevens LJ, van Lipzig MMH, Erpelinck SLA, Pronk A, van Gorp J, Wortelboer HM, et al. A higher throughput and physiologically relevant two-compartmental human ex vivo intestinal tissue system for studying gastrointestinal processes. Eur J Pharm Sci. (2019) 137:104989. doi: 10.1016/j.ejps.2019.104989
171. Shah P, Fritz JV, Glaab E, Desai MS, Greenhalgh K, Frachet A, et al. A microfluidics-based in vitro model of the gastrointestinal human-microbe interface. Nat Commun. (2016) 7:11535. doi: 10.1038/ncomms11535
172. Shim K-Y, Lee D, Han J, Nguyen N-T, Park S, Sung JH. Microfluidic gut-on-a-chip with three-dimensional villi structure. Biomed Microdevices. (2017) 19:37. doi: 10.1007/s10544-017-0179-y
173. Shin W, Kim HJ. Intestinal barrier dysfunction orchestrates the onset of inflammatory host-microbiome cross-talk in a human gut inflammation-on-a-chip. Proc Natl Acad Sci USA. (2018) 115:E10539–47. doi: 10.1073/pnas.1810819115
174. Verhulsel M, Simon A, Bernheim-Dennery M, Gannavarapu VR, Gérémie L, Ferraro D, et al. Developing an advanced gut on chip model enabling the study of epithelial cell/fibroblast interactions. Lab Chip. (2021) 21:365–77. doi: 10.1039/d0lc00672f
175. Kovler ML, Sodhi CP, Hackam DJ. Precision-based modeling approaches for necrotizing enterocolitis. Dis Model Mech. (2020) 13(6):dmm044388. doi: 10.1242/dmm.044388
176. Bossink EGBM, Segerink LI, Odijk M. Organ-on-Chip technology for aerobic intestinal host—anaerobic Microbiota research. Organs-on-a-Chip. (2022) 4:100013. doi: 10.1016/j.ooc.2021.100013
177. Lanik WE, Luke CJ, Nolan LS, Gong Q, Rimer JM, Sarah E, et al. Microfluidic device facilitates novel in vitro modeling of human neonatal necrotizing enterocolitis-on-a-chip. bioRxiv (2020) 11.29.402735. doi: 10.1101/2020.11.29.402735
178. Signore MA, De Pascali C, Giampetruzzi L, Siciliano PA, Francioso L. Gut-on-Chip microphysiological systems: latest advances in the integration of sensing strategies and adoption of mature detection mechanisms. Sens Biosensing Res. (2021) 33:100443. doi: 10.1016/j.sbsr.2021.100443
179. Singhal R, Shah YM. Oxygen battle in the gut: hypoxia and hypoxia-inducible factors in metabolic and inflammatory responses in the intestine. J Biol Chem. (2020) 295:10493–505. doi: 10.1074/jbc.REV120.011188
180. Yang W, Yu T, Huang X, Bilotta AJ, Xu L, Lu Y, et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat Commun. (2020) 11:4457. doi: 10.1038/s41467-020-18262-6
181. Cho SX, Rudloff I, Lao JC, Pang MA, Goldberg R, Bui CB, et al. Characterization of the pathoimmunology of necrotizing enterocolitis reveals novel therapeutic opportunities. Nat Commun. (2020) 11:5794. doi: 10.1038/s41467-020-19400-w
182. Lien E, Chow JC, Hawkins LD, McGuinness PD, Miyake K, Espevik T, et al. A novel synthetic acyclic lipid A-like agonist activates cells via the lipopolysaccharide/toll-like receptor 4 signaling pathway. J Biol Chem. (2001) 276:1873–80. doi: 10.1074/jbc.M009040200
183. Tidswell M, Tillis W, Larosa SP, Lynn M, Wittek AE, Kao R, et al. Phase 2 trial of eritoran tetrasodium (E5564), a toll-like receptor 4 antagonist, in patients with severe sepsis. Crit Care Med. (2010) 38:72–83. doi: 10.1097/CCM.0b013e3181b07b78
184. Wipf P, Eyer BR, Yamaguchi Y, Zhang F, Neal MD, Sodhi CP, et al. Synthesis of anti-inflammatory α-and β-linked acetamidopyranosides as inhibitors of toll-like receptor 4 (TLR4). Tetrahedron Lett. (2015) 56:3097–100. doi: 10.1016/j.tetlet.2014.11.048
185. Neal MD, Jia H, Eyer B, Good M, Guerriero CJ, Sodhi CP, et al. Discovery and validation of a new class of small molecule Toll-like receptor 4 (TLR4) inhibitors. PLoS ONE. (2013) 8:e65779. doi: 10.1371/journal.pone.0065779
186. Tayman C, Uckan D, Kilic E, Ulus AT, Tonbul A, Murat Hirfanoglu I, et al. Mesenchymal stem cell therapy in necrotizing enterocolitis: a rat study. Pediatr Res. (2011) 70:489–94. doi: 10.1203/PDR.0b013e31822d7ef2
187. Rager TM, Olson JK, Zhou Y, Wang Y, Besner GE. Exosomes secreted from bone marrow-derived mesenchymal stem cells protect the intestines from experimental necrotizing enterocolitis. J Pediatr Surg. (2016) 51:942–7. doi: 10.1016/j.jpedsurg.2016.02.061
188. Drucker NA, McCulloh CJ, Li B, Pierro A, Besner GE, Markel TA. Stem cell therapy in necrotizing enterocolitis: current state and future directions. Semin Pediatr Surg. (2018) 27:57–64. doi: 10.1053/j.sempedsurg.2017.11.011
189. McCulloh CJ, Olson JK, Zhou Y, Wang Y, Besner GE. Stem cells and necrotizing enterocolitis: a direct comparison of the efficacy of multiple types of stem cells. J Pediatr Surg. (2017) 52:999–1005. doi: 10.1016/j.jpedsurg.2017.03.028
190. Liu J, Miyake H, Zhu H, Li B, Alganabi M, Lee C, et al. Fecal microbiota transplantation by enema reduces intestinal injury in experimental necrotizing enterocolitis. J Pediatr Surg. (2020) 55:1094–8. doi: 10.1016/j.jpedsurg.2020.02.035
191. Wu H, Guo K, Zhuo Z, Zeng R, Luo Y, Yang Q, et al. Current therapy option for necrotizing enterocolitis: practicalities and challenge. Front Pediatr. (2022) 10:954735. doi: 10.3389/fped.2022.954735
Keywords: intestinal development, neonates, prematurity, necrotizing enterocolitis, intestinal epithelium
Citation: Singh DK, Miller CM, Orgel KA, Dave M, Mackay S and Good M (2023) Necrotizing enterocolitis: Bench to bedside approaches and advancing our understanding of disease pathogenesis. Front. Pediatr. 10:1107404. doi: 10.3389/fped.2022.1107404
Received: 24 November 2022; Accepted: 20 December 2022;
Published: 11 January 2023.
Edited by:
Robert Lewis Schelonka, Oregon Health and Science University, United StatesReviewed by:
Tamas Jilling, University of Alabama at Birmingham, United States© 2023 Singh, Miller, Orgel, Dave, Mackay and Good. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Misty Good bWlzdHlnb29kQHVuYy5lZHU=
Specialty Section: This article was submitted to Neonatology, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.